
View sample multiple sclerosis research paper. Browse research paper examples for more inspiration. If you need a health research paper written according to all the academic standards, you can always turn to our experienced writers for help. This is how your paper can get an A! Feel free to contact our writing service for professional assistance. We offer high-quality assignments for reasonable rates.
Introduction
Multiple sclerosis (MS) is an inflammatory-degenerative disease of the central nervous system (CNS). Its primary target is the myelin sheath via a mechanism that has yet to be explained. The degenerative phase causes destruction of axons and neurons. Except for secondary involvement of the facial nerve in its course within the brainstem, and the nucleus of the trigeminal nerve, it does not affect the peripheral nervous system. It was described well over a hundred years ago, but many of its aspects are still not known. Its pathogenesis is poorly understood and its long-term treatment is ineffectual. Much of its curious epidemiology is unexplained and its genetic transmission unknown. MS is the most common disease of the CNS of the young adult. The first symptoms usually appear at ages 20 to 40. The number of MS patients in the United States has been estimated at about 300 000, a prevalence of 100/100 000. This is probably inaccurate for two reasons: it includes as many as 20–25% of people who have been misdiagnosed as having MS based on the misinterpretation of the magnetic resonance imaging (MRI), and it obviously also does not count the substantial number of persons who have the asymptomatic form of the disease.
Academic Writing, Editing, Proofreading, And Problem Solving Services
Get 10% OFF with 24START discount code
The course of the disease is extremely unpredictable and its clinical presentation is variable, but its predilection for certain parts of the CNS, which include the optic nerves, the brainstem, cerebellum, and cervical spinal cord, eventually provide a characteristic constellation of signs and symptoms.
The Classification Of The Inflammatory Demyelinating Diseases
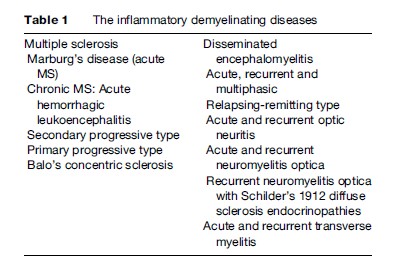
Some neurologists have recently suggested that MS is a part of a spectrum of idiopathic inflammatory demyelinating diseases that differ only in chronicity and severity. For example, acute disseminated encephalomyelitis (DEM) and Marburg’s disease are grouped under the rubric of ‘fulminant,’ whereas Devic’s and Balo’s diseases are included with relapsing myelitis in the group of ‘restricted distribution.’ Such a scheme ignores the pathology of these conditions and seems to have no logical basis. A revised updated classification of the inflammatory demyelinating diseases is presented in Table 1. Marburg’s disease is nothing more than acute MS, as is Balo’s concentric sclerosis, the latter characterized by alternating bands of demyelination. The extremely rare Schilder’s diffuse sclerosis 1912 type has very large lesions. All three of these diseases are often erroneously diagnosed (and treated) as MS because of misread MRIs. Devic’s disease (neuromyelitis optica) is believed to be a variant of DEM (Poser and Brinar, 2004). All the lesions in every condition listed under ‘MS’ in Table 1 exhibit the pathognomonic sharply demarcated edge between normal tissue and demyelination. The various types of DEM differ from MS not only in the absence of the typical plaques but also on the basis of clinical and pathological indices.
Epidemiology of Multiple Sclerosis
Latitude And Prevalence
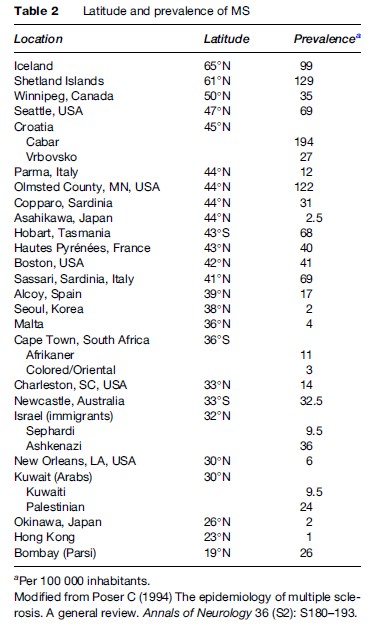
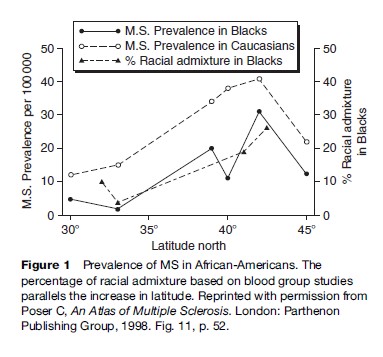
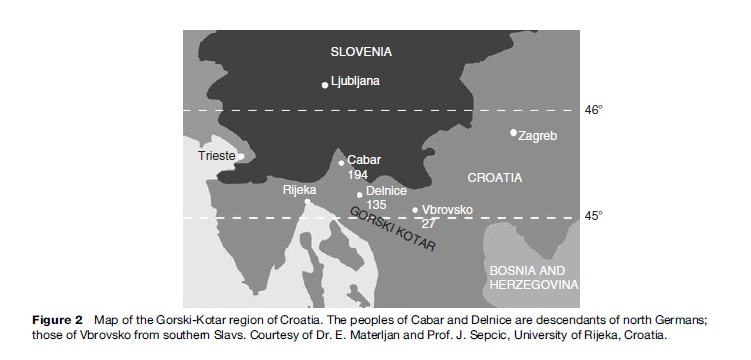
One of the most enduring myths of the epidemiology of multiple sclerosis is that of the direct relationship between latitude and its prevalence. Table 2 makes it clear that prevalence of the disease varies greatly between areas of similar latitude. However, MS prevalence is higher in northern than in southern Europe, as it is in the Americas. This is most likely due to differences in the ethnicity of the populations: MS is much more frequent in persons of both direct (Swedes, Danes, Norwegians, Icelanders) and indirect Scandinavian descent (English, Irish, Russian, Norman, etc.), including Americans in the northern tier of states, many of whom are of Scandinavian origin. The latitude gradient seen in African Americans in the United States, which parallels that of Caucasians but at somewhat lower level, has been explained by the increasing mixture of Caucasian genetic material with progression to the north (Figure 1) (Poser, 1994). There are also many exceptions to the prevalence/latitude rule that militate against it, as is well exemplified by the data in Table 2 for Croatia, Israel, Kuwait, and South Africa. There is a great difference in MS prevalence between three small villages in the Korski Gotar region of Croatia despite an identical environment (Figure 2). The disease is extremely rare in Hindus living in Mumbai, but not uncommon in Parsis living in the same city. No convincingly documented cases of MS have ever been reported in North or South American Indians, in Samis (Lapps), Eskimos, Australian Aborigines, Maoris, Melanesians, Micronesians, or Polynesians. The disease occurs much less frequently in Orientals and is extremely rare in black Africans. There is one well-publicized example of a latitude gradient that has defied explanation: the prevalence of MS in Tasmania is twice that of southern Australia, and has remained that way for 30 years despite a lack of discernible ethnic difference between the populations.
Several reports claim that MS is increasing in frequency. It is hard to know whether this is a true increase or whether it is due to a combination of factors, such as greater awareness of the disease in physicians and the general population, and greater use (and misuse) of imaging. Longer life expectancy and earlier diagnosis may have added to the number of cases, but it is also possible that the disease has in fact become more common.
Genetics
The striking differences in prevalence in similar environments strongly suggest an important role for genetic factors. There is a familial occurrence rate of about 15%. The age-adjusted risk is higher in siblings (3%), parents (2%), and children (2%) than in second and third-degree relatives. There is only a 35% concordance in monozygotic twins, but it is the same as in siblings for dizygotic pairs. Children of conjugal pairs with MS have a much higher prevalence rate (20%), but adopted offspring or other nonbiological relatives have no increased risk (Sadovnick, 1994).
Because of these indications, numerous studies of genetic markers have been carried out, but to date few secure candidates or regions have been identified. Many studies of the class II major histocompatibility complex (MHC) alleles of the human leukocyte antigen (HLA) system and their genotypes have been carried out, but at the family level have been disappointing. Some of these have suggested an association between the MHC alleles DR15 and DQ6 (DRB1* 1501 and DBQ2 *0602) and the gene for tissue necrosis factor encoded within the same linkage group. A specifically different association (with DR4 and its DRB 1*O405-DQA1 *0301-DQB1*0302 genotype) is seen in Mediterranean populations, primarily Sardinians. There is strong evidence indicating that MS is a polygenic disease.
The Vikings And The Genetic Origin Of MS
The highest prevalence rates for MS are found in Iceland, Scandinavia, the British Isles, and the countries settled by their descendants. This suggests that the Vikings may have been instrumental in disseminating the genetic susceptibility to the disease (Poser, 1994a). The Norwegian and Danish Vikings raided in most European countries and settled in large numbers in Normandy (‘the land of the Northmen’), Sicily, and southern Italy. They also founded Dublin in Ireland and York in England. They traveled west and settled in Iceland and the Faroe Islands. The Swedish Vikings originating in Gotland went to the southeast, along the river routes to the Caucasus and the Black and Caspian Seas, and penetrated into Persia, India, and as far as China. They founded Kiev and Novgorod and established Ukraine and the Russian state. Called ‘Varangians,’ they were active in all the military activities of the Byzantine Empire. As early as the tenth century, the Vikings engaged in trade with the Arabs; Arab coins dating from that period have been found in tombs in Iceland and Norway. Vikings from Scandinavia and Iceland, as well as their descendants from England and France, were Crusaders. In fact, the Palestinians born and raised in Kuwait, who have an unexpectedly high prevalence of MS, originated in what had been part of the Latin Kingdom established after the First Crusade. Russians from the Ukraine, and thus of Viking origin, settled in a separate community near what is now Beijing, China, and formed an elite regiment of the Mongol army that participated in the conquests of western Asia and much of southeastern Europe, reaching the gates of Vienna in the thirteenth century. Although the Vikings had a fearful reputation as raiders and plunderers, most of them settled in the areas they had conquered and eventually were integrated into the local population. The practice of capturing and keeping local women as wives or concubines, or selling them as well as other Vikings as slaves, mostly to Arabs, were important factors in this genetic dissemination throughout the world.
Environmental Factors
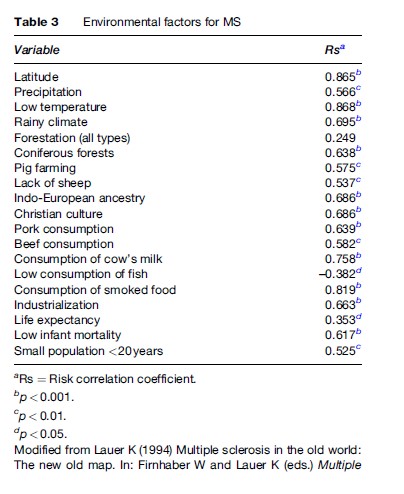
Genetics alone cannot account for the differences in MS observed in various situations; therefore, it is likely that some environmental factors also influence the acquisition of the disease. Many studies of an enormous variety of possible agents and factors that could influence the acquisition or development of the disease have been carried out in various countries, with almost no results. Most of the agents and factors are meaningless, since they are completely lacking in biological plausibility (Table 3) (Lauer, 1994). Usually these factors are related to the clinical onset rather than the acquisition of MS.
Ultraviolet Rays: A Protective Factor?
In recent years, some investigators have focused attention on the intensity of sunshine and ultraviolet rays as possible protection against the acquisition of MS, pointing to the very low prevalence or total absence of the disease in African Blacks, Australian Aborigines, and other highly melanotic ethnic groups. This overlooks the fact that the disease is also completely unknown in non or lightly pigmented groups such as Samis, Inuits, and Eskimos, and that their heavy clothing would effectively screen out the supposedly protective ultraviolet rays. There is no question that MS is extremely rare in African Blacks, but there have been at least four autopsy-proven cases in Senegal. In South Africa, most neurologists are still unwilling to accept the diagnosis in their black patients, thus making the disease seem even rarer.
Migrations
The influence of environmental factors is most apparent in considering the effects of migration on prevalence and incidence rates. Frenchmen living in Africa have a considerably lower prevalence rate than those living in France. The children of West Indian and Asian immigrants to the United Kingdom have the same incidence and prevalence rates as native-born Englishmen, and the Israel-born children of both Ashkenazi and Sephardic Jews also have the same prevalence rates, although their parents’ rates are quite different. Most Martinican Blacks who developed MS had spent significant periods of time in metropolitan France before acquiring the disease. The situation in Hawaii illustrates what appears to be a unique, contradictory situation in which the presumably same environment exerts opposite effects on different ethnic groups (Alter et al., 1971). For persons of Japanese extraction living in Hawaii or in California there is an increased risk of MS compared with those living in Japan (6.5 vs. 2.1); for Caucasians raised in Hawaii it appears to offer some protection against MS (10.5 vs. 34.4). It is difficult to conceive of environmental factors having such a disparate effect unless the genetic make-up of the individual also plays a role in the equation. The existence of a premorbid genetic marker such as the MS ‘trait’ could provide an explanation. Epidemiological studies have demonstrated the primary importance of genetic factors modified by an as yet unrecognized environmental one.
The MS ‘Trait’: A Premorbid Marker Of Genetic Susceptibility
The remarkably low rate of concordance of MS in monozygotic twins has never been fully explained, but it indicates the possibility of a systemic condition called the MS ‘trait’ (MST) (Poser, 2006), which is quite different from asymptomatic MS and may never develop into the disease. It results from the action of an antigenic challenge to the immune system of a genetically vulnerable person that does not cause damage to the nervous parenchyma. A subsequent environmental viral-antigenic event in some MST-carriers can change the trait into the disease. This event could be an infection, which need not be symptomatic, or a vaccination. The MS may become symptomatic, remain asymptomatic, or be manifested only by lesions visible by MRI. It is likely that the development of the MST, called ‘activation,’ occurs early in life, whereas the transition from MST to MS, called ‘acquisition,’ takes place at puberty in most patients, when the immune system is made more vulnerable by the outpouring of female sex hormones. Differences in prevalence between prepuberal migrants, the locally born children of migrants, and their population of origin may also be explained by the MST. Thus, the Hawaii-born Japanese who carries the MST develops MS when he is exposed to an antigenic challenge that is not present in Japan; but the MSTbearing Caucasian does not encounter the appropriate viral antigen, which does not occur in Hawaii, and never develops MS.
Etiology of Multiple Sclerosis
Multiple sclerosis has been described as a disease of unknown etiology, implying the existence of a single cause. A number of infectious agents have been reported as potential etiological agents. They include the corona, measles, Epstein-Barr, herpes simplex type 6, and canine distemper viruses; the human T-cell lymphotrophic virus (HTLV)-l, an ‘MS-associated agent’; and, most recently, chlamydia. None of these has been confirmed, but the idea lingers on, despite exhaustive searches by competent investigators using sophisticated techniques. Nevertheless, so-called ‘epidemics’ in Iceland and in the Faroe Islands continue to be cited as evidence of the infectious nature of the disease, its introduction to the islands ascribed to the arrival of asymptomatic British troops in 1941. The data show a peak of clinical disease onset starting in 1941 and purport to find a direct relationship between the occurrence of MS in natives and British soldiers billeted in their homes. These studies unfortunately are based on the meaningless date of clinical onset; recalculation of the data using the putative date of acquisition at puberty (age 14) clearly show a peak before 1941.
It is important to differentiate between age of clinical onset and age of acquisition of the disease for all etiological and epidemiological investigations. Clinical onset simply means the appearance of symptoms of an already existing disease. The putative date of acquisition of MS has been generally accepted as being at puberty in most if not all patients. This was deduced from the study of MS in English immigrants to South Africa, noting that the disease rarely developed in those who had immigrated before age 15, compared to the number that would have developed it in England (Dean and Kurtzke, 1971). Similar studies confirmed this observation. In support of this concept, and because MS is almost twice as common in women, is that the secretion of female sex hormones, which play an important role in enhancing immune responses, significantly increases in both girls and boys at puberty. Furthermore, many new environmental factors that may influence disease acquisition may be encountered at the same time, including new school, sport, and social activities.
From all of the information that is currently available, it is much more likely that MS is the result in a genetically susceptible subject of the activation of the immune system by different viral agents, which initiates a pathogenetic cascade that is not fully understood and which eventually leads to the destruction of the myelin sheath and the axon.
Pathogenesis of Multiple Sclerosis
Much of what is known about the pathogenesis of multiple sclerosis comes from studies of experimental allergic encephalomyelitis (EAE), an imperfect model of MS but the equivalent of human DEM. In common with EAE, a major alteration of the blood–brain barrier (BBB) is, if not the first, certainly an early and obligatory step in the development of the MS lesion, but EAE does not lead to the formation of the characteristic plaques of MS. In fact, the BBB appears to be injured quite early in the course of illness. Inflammation of capillary and venule walls can be seen in the normal-appearing white matter (NAWM), but it is insufficient to lead to leakage of water and immunoactive substances into the brain parenchyma.
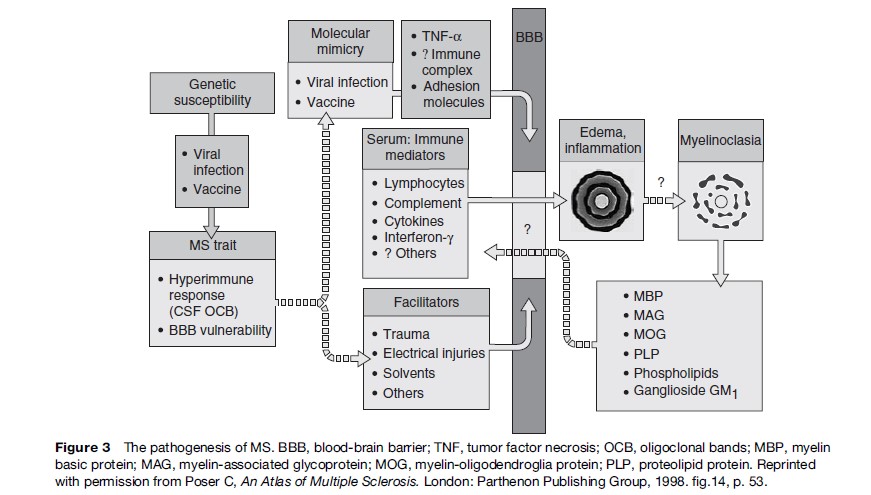
In its early phase MS has several of the features of an autoimmune disease. Although the immune system of MS patients has many abnormalities, the exact pathogenesis remains obscure, and the various schemes that have been proposed are a mixture of fact and speculation. It is unclear whether the immunological alterations are part of the pathogenetic cascade or its results. Ebers (1998) put it succinctly:‘‘It is sobering to realize that despite decades of cellular immunology research of the survey type, no specific abnormality that characterizes or defines MS has been identified.’’ The stimulus for this immunological response is probably an antigenic challenge from an infection, or possibly a vaccination, involving the phenomenon of molecular mimicry. (This is a phenomenon in which some component peptides of the active antigenic molecule are immunologically indistinguishable from a myelin antigen, and hence an appropriate response to infection produces an inappropriate action against some component of the myelin sheath.) A key role has been suggested for tumor necrosis factor, immune complexes, and adhesion molecules, the last named in particular, in the loss of impermeability of the BBB. As a result of the alteration in the BBB, immunoactive T-lymphocytes penetrate into the brain parenchyma. Other immunoactive substances in serum, including complement and interferon-gamma, as well as B-lymphocytes and macrophages, also cross the now permeable BBB and by a still unknown mechanism attack the oligodendroglial–myelin complex. Various cytokines secreted by T-cells have been proposed as the agents of myelinoclasia. The role of the oligodendrocyte in the pathogenetic cascade also remains in dispute, and many consider this rather than the myelin sheath to be the primary target of the process.
The primary effects of MS are inflammation and edema. Myelin destruction does not necessarily follow. Spontaneous resolution of the inflammation and edema without destruction frequently occurs, and provides a logical explanation for the very short duration of some symptoms. Remyelination occurs even in the earliest lesions, but is generally relatively inefficient and much too slow to account for clinical improvement within only a few hours or days. Another explanation may be the activation of alternative or supplementary physiological pathways.
After myelin is destroyed, it is replaced by a glial scar. It is such scars that have given MS its name. In addition to the probable role of the T-cell in causing the inflammatory reaction, antibodies against myelin components also play a crucial role in pathogenesis. The destruction of myelin releases a number of its structural components, including cholesterol, fatty acids, myelin basic protein, myelin-associated glycoprotein, myelin-oligodendrocyte glycoprotein, proteolipid protein, phospholipids, cerebrosides, sphingomyelin, and gangliosides. These substances may enter the bloodstream via the permeable BBB and, in turn, provoke an immune response from systemic lymphocytes, thereby causing a vicious cycle that results in a self-perpetuating condition. This may explain the intermittent progression of the disease. In MS, periods of immune activity, probably stimulated by nonspecific viral infections, are believed to alternate with periods of immunoquiescence. The current status of our understanding, or lack thereof, of the role of the immune system in MS has been summarized by Cedric Raine (1994) as follows:
In sum, while no single immune system molecule can he assigned as unusual to the CSF of MS, and, while there appears to he nothing unique about the manner in which the CNS responds to inflammation, the true uniqueness of the situation in MS is probably related to the many normally sequestered, specific antigens within the myelin sheath and the biology of the myelinating cell, the oligodendrocyte.
A generalized pathogenetic scheme is depicted in Figure 3.
Pathology of Multiple Sclerosis
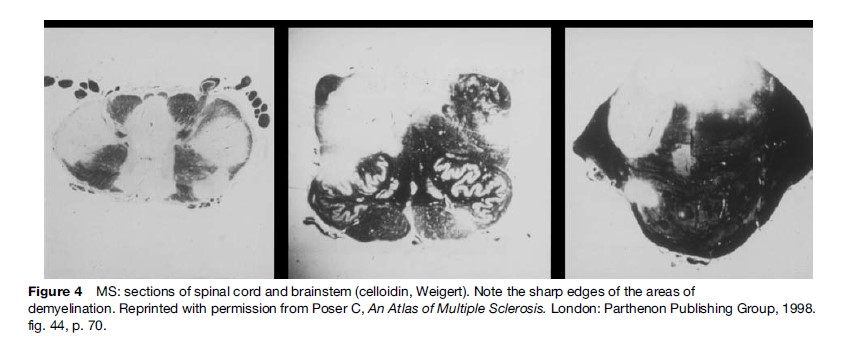
The pattern of destruction of myelin is unique to multiple sclerosis and is seen in none of the other demyelinating (e.g., DEM) or dysmyelinating (e.g., metachromatic or adrenoleukodystrophy) diseases: it consists of plaques that are sharply demarcated from the normal white matter surrounding them (Figure 4). They have been aptly described as ‘‘cut out with a cookie cutter.’’ In contrast, the inflammatory lesions in DEM are almost invariably perivascular and often become confluent. Unless the pathognomonic sharp edge of the MS lesion is captured by the biopsy, it may be impossible to differentiate it from DEM.
Plaques are most commonly seen in the optic nerves and chiasm, the periventricular centrum semiovale, the brainstem, the cerebellar hemispheres, and the cervical spinal cord. Although most plaques are seen in the white matter, they may involve the subcortical U-fibers and extend into gray matter. The cortex, thalamus, the basal ganglia, and the dentate nuclei may all be affected. Lesions of the sensory nucleus of the trigeminal nerve, the intraparenchymal portion of the facial, and some of the connections of the acoustic nerve are frequently involved, but peripheral nerves are spared. Asymmetry of the lesions is the rule.
Inflammatory cellular infiltrates and edema are almost invariably seen in the walls of small blood vessels and the surrounding parenchyma at the edges of the plaques. Similar changes in the walls of capillaries and venules can also be seen in the NAWM. One of the earliest changes is separation of the myelin lamellae by vesicular edema, fragmentation of the sheath, invasion by macrophages that engulf the myelin debris, and eventual denudation of the axon. Most of the plaques show periaxile demyelination, the axon appearing intact, but older lesions clearly show axonal and neuronal degeneration. Clumps of large abnormal gemistocytic astrocytes may be seen near the lesions; these are occasionally mistaken for astrocytomas.
The MS variants, Marburg’s acute MS, Balo’s concentric sclerosis, and Schilder’s 1912 type diffuse sclerosis, all exhibit the same pathological features (Poser and Brinar, 2004).
Physiology and Multiple Sclerosis
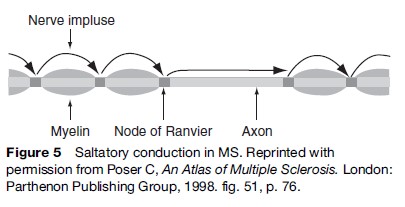
Normal motor and sensory function depend upon the rapid propagation of the nerve impulse along myelinated nerve fibers, measured in milliseconds. The myelin sheath is interrupted at regular intervals by the nodes of Ranvier, where the axon is denuded. Because the axon has a high resistance to the electrical impulse, which makes the speed of conduction too slow, an alternative mechanism takes over. It is called ‘saltatory conduction,’ in which the electrical impulse jumps from one node of Ranvier to the next while achieving the required conduction velocity. However, if the distance between the available nodes is too great because of destruction of some myelin segments, the impulse cannot bridge the gap, and saltatory conduction is no longer possible. The electrical impulse must then travel via the slow axonal route (Figure 5). Once the axon itself is destroyed, conduction is obviously no longer possible and the deficit, if any, becomes permanent.
In some MS patients signs or symptoms appear because nerve conduction slows when body temperature is elevated as a result of either ambient heat or fever. The latter is a common cause of pseudoexacerbations. A body temperature increase of as little as 1 C may be sufficient to cause such signs and symptoms, which disappear upon cooling. Until recently, this was used diagnostically by means of the hot bath test.
Clinical Aspects of Multiple Sclerosis
The Clinical Course
Several clinical types have been recognized: relapsingremitting, primary, and secondary progressive. However, the pathological end result, that is, the sharply bordered plaques of demyelination, is noted in all of them. Some investigators believe that these clinical types represent different diseases or genetic variants of MS, but it is much more likely that the differences in evolution indicate the aggressivity of the disease process, the patient’s susceptibility, and the accumulation of lesions in eloquent areas of the CNS. Many multiple sclerosis lesions remain silent, and a number of routine autopsy series have shown that asymptomatic MS may be as common as the diagnosed condition, with a prevalence of 100/100 000.
Signs And Symptoms
Table 4 summarizes the symptoms of MS at onset in six series, in three of which the diagnosis was confirmed at autopsy. The disease is almost twice as common in women as in men, and the clinical onset is most frequent in the third and fourth decades. Certain neurological complaints in a person under the age of 40 are tantamount to making the diagnosis of MS: a Lhermitte symptom (tingling going down the back when flexing the neck), trigeminal neuralgia, hemifacial spasms, unilateral intention tremor, or binocular diplopia which disappears when closing either eye. Similarly, certain abnormalities of the neurological examination of a young person are also common enough in MS to be of diagnostic value: temporal pallor of the optic disk, unilateral hyperreflexia and a Babinski sign, a significant decrease in position and/or vibratory sensation at the ankles, or some dysmetria on finger-to-nose testing. Dating the clinical onset of MS is often important in epidemiological studies and clinical trials; many patients – and physicians – ascribe symptoms to MS that are unrelated. Table 5 lists the only signs and symptoms that should be considered for that purpose. MS patients may well suffer from headaches, positional vertigo, seizures, back and neck pain, for example, but such signs and symptoms are not part of the MS syndrome. In addition to signs and symptoms indicating involvement of different parts of the CNS, or dissemination in space, and dissemination in time (symptoms appearing at different times) are characteristic of about 2/3 of patients. True exacerbations or bouts appear spontaneously or may follow some kind of viral infection but must be differentiated from pseudo-exacerbations that result from fever, elevated ambient temperature, or some metabolic derangement.
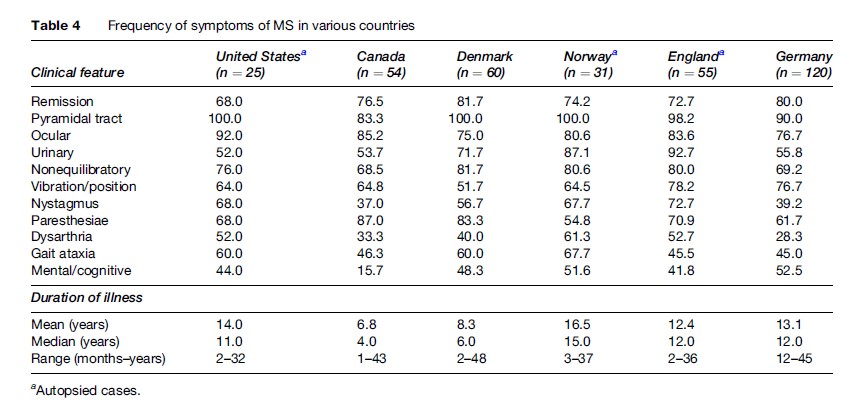
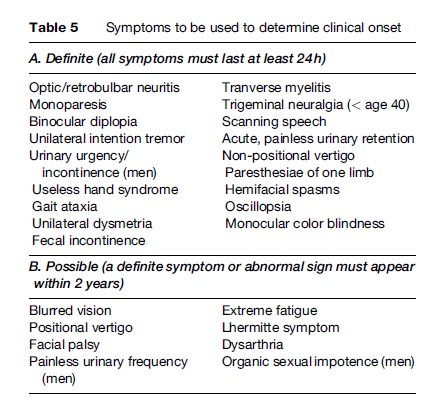
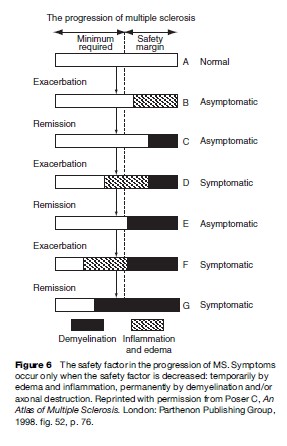
The correlation between the number, site, and size of MS lesions, as revealed by neuroimaging and at autopsy, and clinical manifestations is poor. Many plaques involve so-called ‘silent’ areas of the brain. Furthermore, the disease process must impair conduction in a critical number of fibers in order to produce neurological dysfunction. The available fibers in the affected tract above this number constitute the safety factor. If the signs and symptoms are due to inflammation and edema only, which is almost always the case at the onset of a bout, they will be reversible; it is only when the safety factor is breached, that is, when the number of demyelinated or destroyed fibers exceeds the required minimum, that signs and symptoms become permanent. Figure 6 depicts the mechanism that explains the relapsing-remitting course that develops into secondary progression.
Clinically Isolated Syndromes Suggestive Of MS
Many clinicians now contend that the first manifestation of MS often is optic neuritis, transverse myelitis, or a cerebellar syndrome. Rather than waiting for the confirmatory second episode and fulfilling the classic criteria of dissemination in time and in space, specific, expensive, lifelong treatment is initiated on such flimsy evidence.
Diagnosis of Multiple Sclerosis
Diagnostic Criteria
Because multiple sclerosis is such a variable disease, many diagnostic schemes incorporating a variety of confirmatory tests and procedures have been proposed, but none are accurate and specific enough to eliminate the main differential diagnostic considerations. This applies to the McDonald et al. scheme which is widely used (2001). Although it relies heavily on magnetic resonance imaging (MRI), it still endorses the traditional principles of dissemination in both time and space as the basic diagnostic characteristics.
Among its improvements over previous schemes are the following: the first criteria for primary progressive MS, the recommendation that brainstem auditory and sensorimotor evoked potential studies no longer be considered of diagnostic value, retaining only the visual ones; and most important, the notation that spinal cord lesions should be at least 3 mm but not exceed the length of two vertebrae. The McDonald et al. scheme also warns against forming the diagnosis on the results of a brain biopsy. Unfortunately, however, this scheme is heavily dependent on MRI, for which a series of quantitative measures are offered, in the complete absence of descriptive, qualitative images. These MRI criteria were not derived from a series of clinically well-established cases of MS, but rather on the retrospective review of the MR images of patients who had a clinically isolated syndrome (CIS) and subsequently suffered from a second episode, thus presumably establishing the diagnosis of MS.
The diagnosis of MS remains a clinical one, and usually a careful history coupled with the neurological examination by an experienced neurologist will be sufficient and not require confirmatory procedures. It is regrettable that in the last few years the diagnosis of MS has been based almost exclusively on the interpretation of the MRI by the radiologist. Not only is the latter not necessarily specially trained or experienced but often has been given only a minimum of clinical information.
Examination Of The Cerebrospinal Fluid
Examination of the cerebrospinal fluid (CSF) as an adjunct for the diagnosis of MS has become increasingly rare but remains an essential procedure when other conditions such as Lyme disease, sarcoidosis, HTLV-I-associated paraparesis, AIDS, and neurosyphilis must be ruled out. Thus it remains a useful confirmatory test for MS. The CSF is usually acellular, and the total protein level normal. A protein level above 75 mg% and a white cell count above 15 should raise serious doubts about the diagnosis of MS. Measurement of the level of IgG is important. The simplest and most reliable measurement is the percentage of total CSF protein: less than 15% is considered normal. However, an elevated CSF IgG is nonspecific. It is often observed in many other conditions affecting the nervous system. A more useful examination is the search for oligoclonal bands in the gammaglobulin fraction of protein. To be significant, there must be at least two bands, and none in a coincidental immunoelectrophoresis of the patient’s serum. These bands are present in over 90% of cases and do not vary with disease activity or lack thereof, but are not MS-specific and may be noted in other conditions, including disseminated encephalomyelitis. In the latter, however, they may disappear in time, which never happens in MS.
Visual Evoked Potential Studies
Pattern-reversal visual evoked responses are particularly useful in identifying optic nerve and chiasmatic lesions in patients who have had no symptoms or signs of involvement of the visual system, because they may be delayed in 75% of such patients, including those with normal visual acuity. The critical measurement is that of the peak, designated as P100. The amplitude of the response is of little portent. Interocular differences in P100 delay are usually meaningless. Because the response is modified by changes in visual acuity, it is imperative that the patient wear prescribed corrective lenses during the test. Delay in P100 is far from specific for MS lesions of the optic system; in addition to poor fixation and changes in visual acuity, many other conditions may give false-positive results. Among these are glaucoma, alcohol ingestion, cerebrovascular disease, spinocerebellar degeneration, all types of optic atrophy, and the use of many commonly prescribed drugs.
Neuroimaging
Computed Tomography
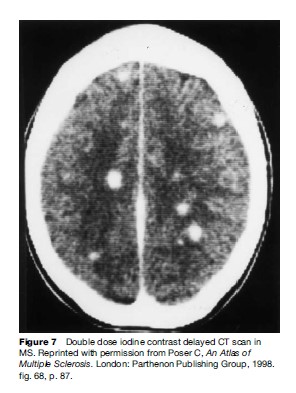
Despite the general availability of MRI in most countries, computed tomography (CT) scanning will undoubtedly remain for many years to come the only neuroimaging procedure available in the poorer areas of the world. Doubling or tripling the dose of intravenous iodinated contrast medium and delaying imaging for one or two hours have greatly enhanced the ability of CT to reveal MS lesions (Figure 7).
Magnetic Resonance Imaging
The introduction of magnetic resonance imaging (MRI) has revolutionized the diagnostic process of MS, but it has proved to be a mixed blessing. Only too often today, the clinician will ignore the history and neurological abnormalities in favor of the radiologist’s report. Approximately 5–15% of clinically definite MS patients have normal MRIs on repeated examination. The correlation between the number, site, and size of MRI white-matter areas of increased signal intensity (AISI) and the clinical signs and symptoms of MS is unreliable. The often-used term ‘burden of disease,’ based on the number and size of lesions, is misleading, as many AISIs may be seen that have persisted for years in clinically normal subjects.
Attempts to establish reliable MRI diagnostic criteria have largely been unsuccessful, because the pattern and characteristics of images associated with MS are nonspecific and are also seen in many other diseases. No pattern of lesions, including the ovoid and perpendicular periventricular lesion, is specific enough to be diagnostic of MS.
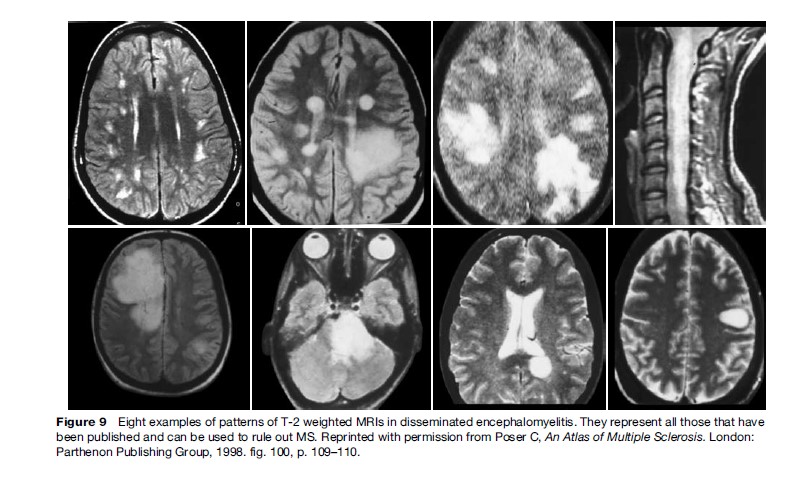
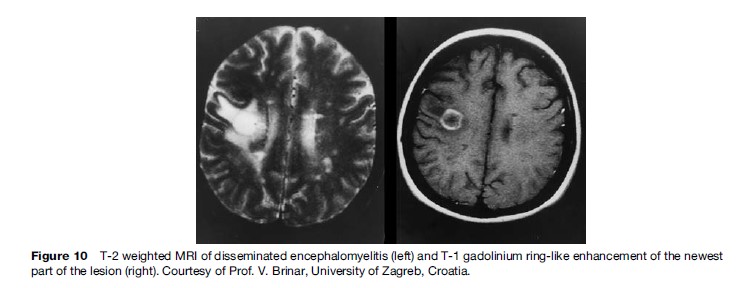
The MRI guidelines that are available are all quantitative; they consist of numbers of lesions without any indications of the details, location, distribution, or size of the images. As new treatments become available, accuracy of diagnosis assumes even greater importance, and correctly distinguishing between MS and other demyelinating conditions producing AISIs on T2-weighted MRIs becomes crucial. By far the most common problem in differential diagnosis is posed by DEM, which may be of the recurrent or polyphasic type and indistinguishable from MS in fulfilling the classical criteria of dissemination in time and in space. None of the published clinical and radiological diagnostic criteria make it possible to differentiate between MS and DEM. Characteristic illustrative MR images of DEM are available: the eight patterns in Figure 9 represent essentially all those that have been published (Poser, 1994). MR images of DEM have often been erroneously misinterpreted as Marburg’s or Balo’s disease, or as brain tumors that have been subjected to the risks of a biopsy. Another helpful differential point is that in DEM ring-like gadolinium enhancement will be seen for almost all the lesions, whereas in MS very few of the AISIs will enhance (Figure 10).
Treatment of Multiple Sclerosis
Symptomatic
Effective drugs have been available for a long time for the treatment for some of the typical problems such as spasticity, the control of urinary urgency and frequency, erectile dysfunction, the pain of trigeminal neuralgia, and the characteristic and disabling severe fatigue. Both oral and intravenous corticosteroids have successfully shortened relapses in many patients (Poser and Brinar, 2002).
Specific
A new era of specific therapy was introduced about ten years ago with the use of the immunomodifying parenteral drugs b-interferon and glatiramer acetate and, to a limited extent, the immunosuppressant mitoxanthrone. These drugs have several possible modes of action on the immune system, but the exact mechanisms of action in MS have not been identified. Despite their side effects, their high cost, and the tendency of some of them to produce antibodies, these lifelong treatments have been used with unbridled enthusiasm and, unfortunately, in some patients whose MS diagnosis was based entirely but erroneously on the MRI interpretation. Although there is convincing evidence that the immunomodulatory and immunosuppressant drugs produce statistically significant decreases in both the number of relapses and of new lesions on MRI, they do not modify the long-term course of the disease (Confavreux et al., 2000).
Another result of the availability of these medications is the practice of initiating treatment after a CIS, such as a single episode of optic neuritis, myelitis, or an isolated cerebellar dysfunction. Rather than wait for a confirmatory second clinical bout, MRI criteria of dubious validity are used to sustain the diagnosis of MS, with the justification that treatment should be started as soon as possible to prevent damage. The fact that the disease has almost always been present for 10 or 15 years is ignored. A great many clinical trials of potential therapeutic agents are in progress. Much attention has been devoted to monoclonal antibodies and to oral tolerization as potential therapeutic agents, but so far results have been disappointing.
Support Systems
Many MS sufferers pursue productive and enjoyable lives despite their disease. Patients should be encouraged to continue working as long as possible or to seek accommodations to their limitations. Participation in sports, even if confined to a wheelchair, is possible; horseback riding is popular with MS patients. Also available are wheelchair basketball teams and ‘sitdown’ skiing. People with MS must be able to depend upon their immediate families for moral support. In many places however, there are specialized MS clinics that offer comprehensive care, including medications, psychiatric and social service counseling, and physical and occupational rehabilitation. Some patients belong to peer groups whereas others avoid them; in some cities, there are day centers run by the local MS society. Other services that may be available are transportation and the loan of wheelchairs and walkers. Wheelchair-bound patients require access to restaurants, museums, theaters, and other public facilities via ramps and elevators, but many cities have yet to recognize this need.
Bibliography:
- Alter M, Okihiro M, Rowley W, et al. (1971) MS among Orientals and Caucasians in Hawaii. Neurology 21: 122–130.
- Confavreux C, Vukusic S, Moreau T, et al. (2000) Relapses and progression of disability in multiple sclerosis. New England Journal of Medicine 343: 1430–1438.
- Dean G and Kurtzke J (1971) On the risk of multiple sclerosis according to the age of immigration to South Africa. British Medical Journal 3: 725–729.
- Ebers G (1998) Immunology. In: Paty D and Ebers G (eds.) Multiple Sclerosis, p. 410. Philadelphia, PA: Davis.
- Lauer K (1994) Multiple sclerosis in the old world: The new old map. In: Firnhaber W and Lauer K (eds.) Multiple Sclerosis in Europe. An Epidemiological Update, p. 19. Alsbach/Bergstrasse, Germany: Leuchtturm Verlag.
- McDonald W, Compston A, Edan G, et al. (2001) Recommended diagnostic criteria for multiple sclerosis: Guidelines from the international panel on the diagnosis of multiple sclerosis. Annals of Neurology 50: 121–127.
- Poser C (1994) The epidemiology of multiple sclerosis. A general review. Annals of Neurology 36(S2): S180–193.
- Poser C (1994a) The dissemination of multiple sclerosis: A Viking saga? A historical essay. Annals of Neurology 36(S2): S321–343.
- Poser C (2006) The multiple sclerosis trait and the development of multiple sclerosis: Genetic vulnerability and environmental effect. Clinical Neurology and Neurosurgery 108: 227–233.
- Poser C and Brinar V (2002) The symptomatic treatment of multiple sclerosis. Clinical Neurology and Neurosurgery 104: 231–235.
- Poser C and Brinar V (2004) The nature of multiple sclerosis. Clinical Neurology and Neurosurgery 106: 139–171.
- Raine C (1994) The immunology of multiple sclerosis. Annals of Neurology 36(S1): S61–72.
- Sadovnick A (1994) Genetic epidemiology of multiple sclerosis: A survey. Annals of Neurology 36(S2): S194–203.
ORDER HIGH QUALITY CUSTOM PAPER

Always on-time

Plagiarism-Free

100% Confidentiality