
Sample Genetic Determinants Of Obesity Research Paper. Browse other research paper examples and check the list of research paper topics for more inspiration. If you need a religion research paper written according to all the academic standards, you can always turn to our experienced writers for help. This is how your paper can get an A! Feel free to contact our research paper writing service for professional assistance. We offer high-quality assignments for reasonable rates.
1. Background
Obesity is a worldwide epidemic. Thus it has been estimated that about 7 percent of the adults of the whole world have a body mass index (BMI; body weight in kilograms divided by height in meters squared) in the obesity range (i.e., 30 kg/m2 or more). This translates into about 250 millions of obese adults (Seidell 2000). This amounts to about 300,000 deaths per year in the USA alone attributable to obesity (Allison et al. 1996). From a slowly increasing rate of obesity during the 1960s and 1970s, the last two decades have seen an explosion in the rate of increase. A rise of 30 to 50 percent in the prevalence rate between 1980 and 1998 has been noted in many countries (WHO 1998, NHLBI 1998). It is thought that this epidemic reflects an interplay between environmental and genetic influences. Although environmental changes must underlie the rapid progression of obesity in recent years, this explosion is occurring on a genetic background, perhaps one characterized by a vulnerability in the presence of current living conditions. Some populations such as the Polynesians and Micronesians, the Native Americans, and Africans in the diaspora seem particularly susceptible.
Academic Writing, Editing, Proofreading, And Problem Solving Services
Get 10% OFF with 24START discount code
In this research paper we will examine the role that genetics has played in this epidemic. Recently, scientists involved in the study of the cause of human obesity have become optimistic about the possibility of identifying the genes associated with the predisposition to this disease. There are good reasons to believe that this new enthusiasm is justified. A growing understanding of the human genome, the high degree of homology between humans and common laboratory mammal models for a large number of genes and chromosomal regions, and the availability of a whole variety of technologies and tools to study and manipulate DNA in the laboratory are among the most important reasons for the present level of hope in the obesity research community.
2. Current Theory
The animal literature on obesity has produced a useful base for approaching the genetics of human obesity (Bray and York 1998, Kalra et al. 1999). This animal literature teaches us that we can view obesity as a disorder in a normally well-controlled system. There are four components in this system:
(a) The brain, or controller, which receives information from many sources, transduces this information into useful signals and controls food intake, the autonomic nervous system, muscular activity, and metabolism.
(b) The efferent signals by which the brain regulates food intake and metabolism involve voluntary muscle groups, the autonomic nervous system, and the hormonal system.
(c) The controlled system, which includes the organ systems involved with identifying, ingesting, digesting, absorbing, storing, or metabolizing nutrients and the excretion of gaseous, liquid, and solid waste.
(d) The afferent signals, which provide information to the brain, include the sensory organs, vagal and sympathetic nervous systems, circulating hormones like leptin, nutrient signals, and peripherally generated hormones.
This feedback approach can be used to locate the defects that produce significant obesity (Fig. 1).
Several defects in the brain produce obesity. Clinically, at the turn of the twentieth century, Frolich and Babinski described separate cases of hypothalamic obesity tumors that produced obesity (see Bray 1993). ‘Hypothalamic obesity’ can be produced routinely by damaging structures in the medial hypothalamus (paraventricular nucleus, PVN). This is probably the result of damaging areas that are critical to activating inhibitory systems in the medial hypothalamus.
Four genetic manipulations of this system also produce massive obesity. Leptin deficiency or leptin receptor defects impair the control of the afferent signals generated by leptin acting on the arcuate nucleus (ARC in Fig. 1). In the absence of leptin, neuropeptide Y (NPY) increases and proopiomelanocortin (POMC) decreases food intake, a setting that matches the hypothalamic lesion. Genetic overexpression of agouti-related peptide (AGRP), which can block the inhibitory effects of α-MSH produced from POMC by post-translational processing or targeted disruption at the melanocortin-4 receptor, also produces massive obesity (Bray 1998).
The other defects identified in Fig. 1 produce more moderate forms of obesity. Defects in the cholecysto-kininA (CCKA) receptor and in the bombesin-3 receptor produce modest obesity by interfering with afferent signaling targeted disruption of the serotonin 2C receptor. Similarly, overexpression of corticotropin releasing hormone (CRH) or disabling of the glucocorticoid receptor in the hypothalamus produces moderate obesity.
Melanin-concentrating hormone increases food in- take. Targeted disruption of this gene produces lean-ness, suggesting a tonic involvement in feeding and body fat regulation. Two other peptides that increase food intake, NPY and orexin, do not appear to be essential since targeted disruption of peptide production does not affect food intake or body fat. Any of the peptides listed above might serve as the basis for a new drug treatment for obesity.
3. Evidence From Genetic Epidemiology
In a report published by the Carnegie Institute of Washington in 1923, C. B. Davenport (Davenport 1923) described the first comprehensive attempt to understand the role of inheritance in human body mass for stature. His study demonstrated quite convincingly that body mass index (BMI) values were more similar among family members than among unrelated persons. However, he noted that normal weight parents sometimes have obese adult offspring. He also observed the converse: obese parents frequently have normal weight adult descendants. Thus, the genetics of human obesity is likely to be complicated.
Since Davenport’s report, the literature on genetics and obesity has expanded and deepened as a variety of new approaches have come to the fore. The Danish Adoption Registry Study, the Veterans Administration Twins Study, the Quebec Family Study, and several other studies were instrumental in reawakening interest in this problem. An overview of some of these genetic epidemiologic data is presented below.
3.1 Heritability Levels
The level of heritability has been considered in a large number of twin, adoption, and family studies. The level of heritability is simply the fraction of the population variation in a trait (e.g., BMI) that can be explained by genetic transmission. Such studies use a variety of procedures to control or take into account age and sex differences. Results obtained by a good number of investigators indicate that the heritability level estimates depend on how the study was conducted and on the kinds of relatives upon which they are based (Table 1). For instance, studies conducted with identical twins and fraternal twins or identical twins reared apart have yielded the highest heritability levels with values clustering around 70 percent of the variation in BMI.

In contrast, the adoption studies have generated the lowest heritability estimates of about 30 percent or less. The family studies have generally found levels of heritability intermediate between the twin and the adoption study reports. A few investigations have included all or most of these kinds of relatives in the same analysis. Using analytical techniques developed to use all the information and maximum likelihood procedures, these studies have concluded that the true heritability estimate for BMI in large sample sizes was between 25 and 40 percent. Recent surveys undertaken in Sweden and the USA with the collaboration of severely obese and morbidly obese subjects together with information obtained on their parents, siblings, and spouses confirm that the genetic contribution to obesity may indeed be around 25 to 40 percent of the individual differences in BMI (Bouchard et al. 1998).
3.2 Familial Risk For Obesity
The risk of becoming obese when a first-degree relative is overweight or obese can be quantified using the lambda coefficient (λs) defined as the ratio of the risk of being obese when a biological relative is obese compared with the risk in the population at large, i.e., the prevalence of obesity (Risch 1990). Ageand genderstandardized risk ratios (equivalent to λs) obtained from 2349 first degree relatives of 840 obese probands and 5851 participants of the National Health and Nutrition Examination Survey III (NHANES III) revealed that the prevalence of obesity (BMI ≥ 30) is twice as high in families of obese individuals than in the population at large (Lee et al. 1997). Moreover, the risk increases with the BMI threshold used to define the obesity. Thus, the risk of extreme obesity (BMI ≥ 45) is about eight times higher in families of extremely obese subjects.
The familial clustering of morbid obesity was also investigated (Adams et al. 1993) in 221 families (n = 1560, subjects ages 18 years and older) in Utah ascertained through a single morbidly obese proband. All probands had to be at least 45.5 kg over their ideal body weight. About 48 percent of the morbidly obese probands had one or more first-degree relatives who were also morbidly obese. Using data from 20,455 families from the general Utah population, the authors estimated that the prevalence of morbid obesity reached 6 percent in the Utah population. Thus, the risk of morbid obesity was about eight times higher (6 percent vs. 48 percent) in families segregating for morbid obesity than in the general population. More recently, using data from 15,245 participants aged 7 to 69 years from the 1981 Canada Fitness Survey, it was shown that the familial risk of obesity was 5 times higher for relatives in the upper 1 percent of the distribution of BMI than in the general Canadian population (Katzmarzyk et al. 1999).
4. Animal Models
4.1 Single Gene Models
Between the first wave of genetic and family studies before World War II and the second wave beginning around 1985, there was a nearly 50-year hiatus in human studies. Filling this gap was a growing literature on the heritage of obesity in animal studies. Before World War II, only the dominantly inherited yellow-obese mouse had been identified. Following the spontaneous appearance of a recessively inherited form of obesity in mice in 1950, four other recessively inherited forms of obesity were described in mice (Table 2). Two genetic forms of obesity were also found in rats.
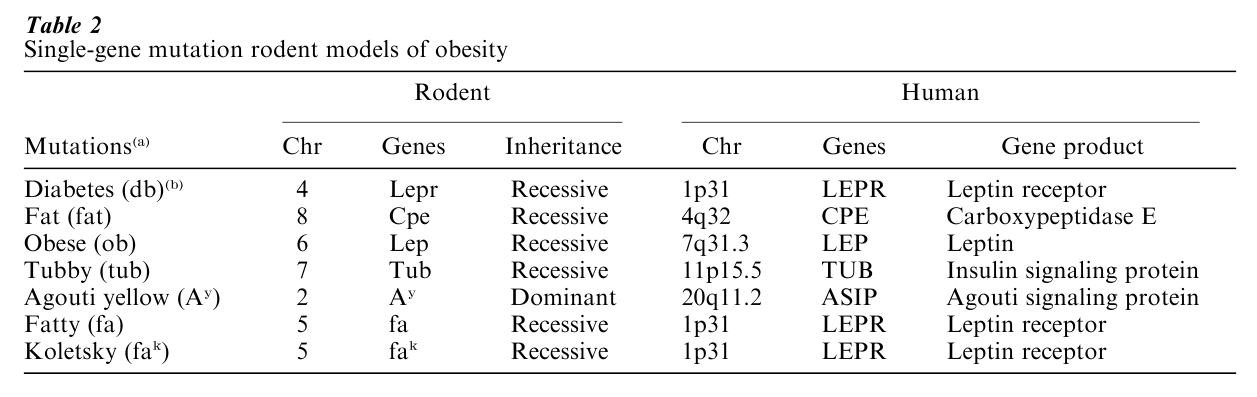
These animal models, through the skepticism of many, have proved to be a boon to the entire field of genetics and obesity. The genetic defect in the yellow-obese mouse unraveled in 1992, and a defect in a similar pathway is now known to produce human obesity in some individuals. The discovery of leptin, the defective product in the obese (ob/ob) mouse, has opened a whole new field of research on the control of energy stores. Leptin deficiency is the cause of obesity in three families described so far, and treatment with leptin reverses the defects. A defect in the leptin receptor causes obesity in mice, rats, and humans. Failure to convert prohormone peptides into their active peptides causes obesity in mice as well as humans. Thus, the lessons from experimental biology have paid rich rewards in the study of human obesity.
4.2 Transgenics
Studies in transgenic animals have added new depth and a few surprises to our understanding of the genetics of obesity (Table 3). This new technology produces two kinds of engineered animals by altering the germ-cell line. One type of transgenic mouse overexpresses a given peptide. One example of obesity produced by overexpression is the yellow-obese mouse. Overexpression of either the agouti protein itself or an agouti-related peptide will produce massive obesity.

The second transgenic strategy is to produce targeted disruption of a single gene—a so-called knockout. Knocking out leptin production or knocking out the melanocortin-4 receptor in the hypothalamus will each produce massive obesity. These engineered rodents have proven to be extremely useful for the dissection of the mechanisms regulating energy balance and for the understanding of the biological pathways leading to leanness or fatness.
5. Human Single Gene Defects
Table 2 Single-gene mutation rodent models of obesity Although individuals affected with Prader-Willi Syndrome, Bardet-Biedl Syndrome, or other Mendelian obesity syndromes represent a very small fraction of the obese population, they reveal that single gene defects can be important in the development of human obesity. The number of cases with single gene mutations causing obesity or associated with it has increased substantially over the past few years, but still represents a very small fraction of all the obesity cases worldwide. There are now 30 cases reported in the literature involving 11 mutations in 7 genes (Perusse et al. 1999, Chagnon et al. 2000). These cases are listed in Table 4 and are reviewed briefly below.
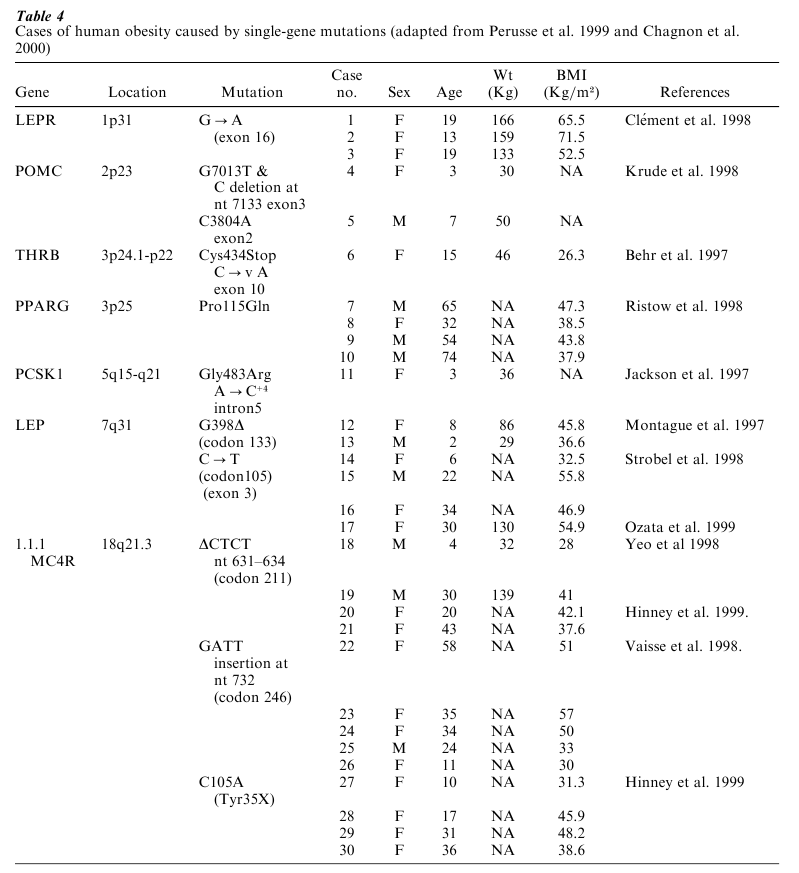
In the leptin receptor (LEPR) gene, a G to A base substitution in exon 16 was found in three severely obese individuals who also showed pituitary dysfunction (Clement et al. 1998). Mutations in the proopiomelanocortin (POMC) gene were found in two patients with severe early-onset obesity, adrenal insufficiency, and red hair pigmentation (Krude et al. 1998). One was a compound heterozygote for two mutations in exon 3, a G to T change at nucleotide (nt) 7013, causing a premature termination at codon 79 and a 1 bp deletion at nt 7133 which also caused a premature termination, but at codon 131. The other subject was homozygous for a C to A transversion at nt 3804 in exon 2. Mild obesity along with combined hypothyroid and hyperthyroid symptoms was reported in a heterozygous patient with a point mutation in exon 10 of the thyroid hormone receptor, beta (THRB) gene, which produced a stop codon with a 28 aminoacid deletion and abolished T3 binding (Behr et al. 1997).
Four markedly obese individuals have been shown to be carriers of a missense mutation in the peroxisome proliferator activated receptor, gamma (PPARG2) gene (Ristow et al. 1998). This Pro to Gln conversion at position 115 was associated with a higher BMI compared with an obese control population without the mutation. One case of an impaired prohormone processing associated with a G to A substitution at codon 483 in the protein convertase subtilisin/kexin type 1 (PCSK1) gene was also reported (Jackson et al. 1997). One of the earliest identified mutations causing obesity was reported by Montague (Montague et al. 1997) in the leptin (LEP) gene. Two severely obese children with a mutation in codon 133 (G398∆) were then identified. Subsequently, three individuals with a mutation (C105T) in exon 3 of the LEP gene were reported (Strobel et al. 1998). Further investigation of the same family yielded an additional female member carrying the same mutation (Ozata et al. 1999).
Two different groups reported on nine patients who were found to have mutations in the melanocortin 4 receptor (MC4R) gene associated with a dominant form of obesity (Yeo et al. 1998, Vaisse et al. 1998). Two heterozygous males shared a CTCT deletion at codon 211 which resulted in a missing leucine and a premature stop codon. Seven other subjects had a GATT insertion at nt 732 resulting in the expression of a nonfunctional truncated receptor. More recently, Hinney et al. (1999) described six female obese subjects with mutations in the MC4R gene, two with the already identified CTCT deletion at codon 211, and four others (two probands and their respective mothers) with a novel mutation at position 35 leading to a premature stop codon generating a truncated protein product. Seven missense mutations in MC4R of unknown significance in seven other extremely obese subjects (BMI 99th percentile) were also described.
6. Scanning The Whole Human Genome
Several other strategies have been used in the effort to identify the genes and mutations responsible for the predisposition to obesity. Among them, one finds a whole series of studies on candidate genes. These have been reviewed elsewhere (Bouchard et al. 1998, Chagnon et al. 2000) and will not be discussed here. In general, however, the results of these observational studies have been disappointing, with small sample size, small effect size, and lack of replication studies or failure to replicate being the most common problems.
Another approach has been that of performing genome-wide scans with a view to identifying chromosomal regions of interest. These studies have typically been performed with pairs of sibs, sometimes whole nuclear families or pedigrees, and are based on about 300 microsatellite markers and more. The results of two such genomic scan studies pertaining to massive obesity are summarized briefly here. They are based on families recruited through a proband with a BMI greater than 40.
The first has been conducted on a French cohort including 514 subjects (264 sib-pairs) from 158 nuclear families. Multipoint linkage analysis using affected sib-pairs revealed significant evidence of linkage with obesity (Lod score = 4.85) on chromosome 10p12 (Hager et al. 1998). The second study was based on a mixed sample of Caucasian and African-American subjects from 124 nuclear families (Lee et al. 1999). The authors tested linkage and found significant evidence of linkage (3.02 ≤ Lod score ≤ 3.16) between obesity and markers located on chromosome 20q13. Other genomic scans for BMI or body fat levels measured variously have also been published from the San Antonio Heart Family Study, the Pima Indian Sibling Study, and the Quebec Family Study, but will not be reviewed here as they focus on less severe obesity.
7. Conclusion
It is quite likely that genetic susceptibility to the more severe forms of human obesity is stronger than for the milder types of obesity or for simple overweight. This is concordant with the observation that the risk of morbid obesity is higher when parental obesity is present and with an early-onset of the disease as shown by a recent study (Whitaker et al. 1997).
Obesity is a complex multifactorial trait evolving under the interactive influences of dozens of affectors from the social, behavioral, physiological, metabolic, cellular, and molecular domains (Bouchard et al. 1998). Segregation of the genes is not easily detected in familial or pedigree studies and whatever the influence of the genotype on the etiology, it is generally attenuated or exacerbated by nongenetic factors. From the research currently available, a good number of genes seem to have the capacity to cause obesity or increase the likelihood of becoming obese. This may be a reflection of how most human obesity comes about. In other words, the susceptible genotypes may result from allelic variations at a good number of genes. There are, however, cases in which a single mutation in a key gene is sufficient to cause obesity. Several such mutations have been uncovered in the past few years and many new ones are likely to be evidenced in the future. However, it must be recognized that the common forms of obesity do not arise as a result of a single mutation. Recent progress in animal genetics, transfection systems, transgenic animal models, recombinant DNA technologies applied to positional cloning, and methods to identify loci contributing to quantitative traits have given a new impetus to this field. Because the peptides and monoamines that are being targeted also have other behavioral consequences, the entire scope of information in the behavioral and social sciences will be enhanced. The stage is now set for major advances to occur in the understanding of the genetic and molecular basis of complex diseases such as human obesity.
Bibliography:
- Adams T D, Hunt S C, Mason L A, Ramirez M E, Fisher A G, Williams R R 1993 Familial aggregation of morbid obesity. Obesity Research 1: 261–70
- Allison D B, Faith M S, Nathan J S 1996 Risch’s lambda values for human obesity. International Journal of Obesity 20: 990–9
- Behr M, Ramsden D B, Loos U 1997 Deoxyribonucleic acid binding and transcriptional silencing by a truncated c-erbA beta-1 thyroid hormone receptor identified in a severely retarded patient with resistance to thyroid hormone. Journal of Clinical Endocrinology and Metabolism 82: 1081–7
- Bouchard C (ed.) 1994 The Genetics of Obesity. CRC Press, Boca Raton, FL
- Bouchard C, Perusse L, Rice T, Rao D C 1998 The genetics of human obesity. In: Bray G A, Bouchard C, James W P T (eds.) Handbook of Obesity. Marcel Dekker, New York
- Bray G A 1993 Commentary on classics of obesity: 4. Hypothalamic obesity. Obesity Research 1: 325–8
- Bray G A 1998 Contemporary Diagnosis and Management of Obesity. Handbooks in Health Care Company, Newton, PA
- Bray G A, York D A 1998 The MONA LISA hypothesis in the time of leptin [Review]. Recent Progress in Hormone Research 53: 95–117 [discussion: 117–18]
- Chagnon Y C, Perusse L, Weisnagel S J, Tankinen T, Bouchard C 2000 The human obesity gene map: The 1999 update. Obesity Research 8: 89–117
- Clement K, Vaisse C, Lahlou N, Cabrol S, Pelloux V, Cassuto D, Gourmelen M, Dina C, Chambaz J, Lacorte J M, Basdevant A, Bougneres P, Lebouc Y, Froguel P, Guy-Grand B 1998 A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature 392: 398–401
- Davenport C B 1923 Body build and its inheritance. Carnegie Institution of Washington, Washington, DC
- Hager J, Dina C, Francke S, Dubois S, Houari M, Vatin V, Vaillant E, Lorentz N, Basdevant A, Clement K, Guy-Grand B, Froguel P 1998 A genome-wide scan for human obesity genes reveals a major susceptibility locus on chromosome 10. Nature Genetics 20: 304–8
- Hinney A, Schmidt A, Nottebom K, Heibult O, Becker I, Ziegler A, Gerber G, Sina M, Gorg T, Mayer H, Siegfried W, Fichter M, Remschmidt H, Hebebrand J 1999 Several mutations in the melanocortin-4 receptor gene including a nonsense and a frameshift mutation associated with dominantly inherited obesity in humans. Journal of Clinical Endocrinology and Metabolism 84: 1483–6
- Jackson R S, Creemers J W M, Ohagi S, Raffin-Sanson M L, Sanders L, Montague C T, Hutton J C, O’Rahilly S 1997 Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nature Genetics 16: 303–36
- Kalra S P, Dube M G, Pu S, Xu B, Horvath T L, Kalra P S 1999 Interacting appetite-regulating pathways in the hypothalamic regulation of body weight [Review]. Endocrine Reviews 20(1): 68–100
- Katzmarzyk P T, Perusse L, Rao D C, Bouchard C 1999 Familial risk of obesity and central adipose tissue distribution in the general Canadian population. American Journal of Epidemiology 149: 933–42
- Krude H, Biebermann H, Luck W, Horn R, Brabant G, Gruters A 1998 Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nature Genetics 19: 155–7
- Lee J H, Reed D R, Li W D, Xu W, Joo E J, Kilker R L, Nanthakumar E, North M, Sakul H, Bell C, Price R A 1999 Genome scan for human obesity and linkage to markers in 20q13. American Journal of Human Genetics 64: 196–209
- Lee J H, Reed D R, Price R A 1997 Familial risk ratios for extreme obesity: Implications for mapping human obesity genes. International Journal of Obesity and Related Metabolic Disorders 21: 935–40
- Montague C T, Farooqi I S, Whitehead J P, Soos M A, Rau H, Wareham N J, Sewter C P, Digby J E, Mohammed S N, Hurst J A, Cheetham C H, Earley A R, Barnett A H, Prins J B, O’Rahilly S 1997 Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature 387: 903–8
- National Institutes of Health and National Heart, Lung, and Blood Institute 1998 Clinical guidelines on the identification, evaluation, and treatment of overweight and obesity in adults: The Evidence Report. Obesity Research 6 (Suppl. 2): 51S–209S
- Ozata M, Ozdemir I C, Licinio J 1999 Human leptin deficiency caused by a missense mutation: Multiple endocrine defects, decreased sympathetic tone, and immune system dysfunction indicate new targets for leptin action, greater central than peripheral resistance to the effects of leptin, and spontaneous correction of leptin-mediated defects. Journal of Clinical Endocrinology and Metabolism 84: 3686–95
- Perusse L, Chagnon Y C, Weisnagel J, Bouchard C 1999 The human obesity gene map: The 1998 update. Obesity Research 7: 111–29
- Risch N 1990 Linkage strategies for genetically complex traits. I. Multilocus models. American Journal of Human Genetics 46: 222–8
- Ristow M, Muller-Wieland D, Pfeiffer A, Krone W, Kahn C R 1998 Obesity associated with a mutation in a genetic regulator of adipocyte differentiation. New England Journal of Medicine 339: 953–9
- Seidell J C 2000 The current epidemic of obesity. In: Bouchard C (ed.) Physical Activity and Obesity. Human Kinetics Publishers, Champaign, IL
- Strobel A, Issad T, Camoin L, Ozata M, Strosberg A D 1998 A leptin missense mutation associated with hypogonadism and morbid obesity. Nature Genetics 18: 213–15
- Vaisse C, Clement K, Guy-Grand B, Froguel P 1998 A frameshift mutation in human MC4R is associated with a dominant form of obesity. Nature Genetics 20: 113–14
- Whitaker R C, Wright J A, Pepe M S, Seidel K D, Dietz W H 1997 Predicting obesity in young adulthood from childhood and parental obesity. New England Journal of Medicine 337: 869–73
- World Health Organization 2000 Obesity—Preventing and Managing the Global Epidemic. Report of a WHO consultation on obesity. World Health Organization, Geneva
- Yeo G S H, Farooqi I S, Aminian S, Halsall D J, Stanhope R G, O’Rahilly S 1998 A frameshift mutation in MC4R associated with dominantly inherited human obesity. Nature Genetics 20: 111–12
ORDER HIGH QUALITY CUSTOM PAPER

Always on-time

Plagiarism-Free

100% Confidentiality