
Sample Depression Research Paper. Browse other research paper examples and check the list of research paper topics for more inspiration. iResearchNet offers academic assignment help for students all over the world: writing from scratch, editing, proofreading, problem solving, from essays to dissertations, from humanities to STEM. We offer full confidentiality, safe payment, originality, and money-back guarantee. Secure your academic success with our risk-free services.
This research paper describes how over the past ten years a solid neurobiological database has emerged that has left no doubt that depression is a complex clinical condition where interaction between genetic endowment and exogenous factors determines the phenotype. The various hypotheses for central nervous system dysfunction that may precipitate depression are examined and the ways in which studies related to stress hormone regulation have led to hypothesisdriven drug discovery strategies are discussed. In the future, the descriptive behavioral aspects of psychiatry will be replaced by more medicallyoriented schemes where abnormal behavior and mood are viewed as the functional readout of impaired gene environment interaction. Thus, traditional diagnostic schemes, presently still entirely based on behavioral and demographic data, will be modified by the new and exciting database that is emerging from functional genomics. Notably, most types of behavior depend on the interplay between environmental factors and multiple genes.
Academic Writing, Editing, Proofreading, And Problem Solving Services
Get 10% OFF with 24START discount code
Based upon the complete genome sequences of humans, the clinical management of depression will become more individualized on the basis of functional phenotyping with laboratory tests and genotyping, once appropriate maps for polymorphisms have been established. The future knowledge base that will include not only behavioral, but also functional and genetic information about the patient will be so powerful that we will wonder how we ever practiced clinically without it.
1. The Socioeconomic Burden Of Depression
Depression is one of the most common medical conditions worldwide. A national survey conducted in the United States of America has shown that the average 12-month prevalence in the general population is 10.3% with a 1-month prevalence ranging between 1.5% and 2.5%, amounting to a lifetime prevalence of 17% (Kessler et al. 1994). New research has also led to the abandonment of the view that depression is a byproduct of stressful life styles in industrialized countries. In fact, studies conducted under the auspices of the World Health Organization (WHO) indicate that the situation may be even worse in poor countries where malnutrition and infection make the brain more susceptible to mental disorders. Notably, five out of the ten leading causes of disability worldwide are psychiatric conditions. Using a sophisticated method for assessing the duration and severity of a disability, disability-adjusted life years (DALY), it has been calculated that depression will become a leading cause of disability in the coming decades, second only to ischemic heart disease (see Table 1).

It is also abundantly clear that one consequence of depression is a fourfold risk of developing ischemic heart disease and further, that patients who develop depression following a myocardial infarction, have a 15% higher mortality rate within the first six months following the attack than those without depression.
One of the most dramatic sequelae of depression is suicide. Official suicide rates drastically underestimate the actual level of suicide by varying extents. According to conservative estimates suicide claims over a million deaths per year, and among the younger population suicide is a leading cause of death second only to accidents. Given these facts and focusing on prevalence rates, it needs to be emphasized that these rates represent only diagnosed cases, and therefore only a fraction of the total prevalence because depression is frequently overlooked but precedes suicide in the majority of cases. It is estimated that only approximately 50% of the population with depression in Europe and the United States is correctly diagnosed, while in Japan the rate is much lower, not exceeding 20%.
As a consequence, depression is still widely undertreated and remains one of medicine’s most costly illnesses. When combining direct costs such as inpatient and outpatient care and pharmaceutical or talk therapies with the indirect costs of lost productivity, missed work days and loss of lifetime earnings, it becomes clear that depression accounts for a major share of the overall health care expenditure. These costs were calculated to amount to US$ 43 billion in 1990 in the United States alone, and a realistic reflection of the economic size of the burden imposed by depression worldwide underscores the pressing need to increase efforts on all fronts, ranging from public awareness to pharmaceutical research. In the light of these figures and the fact that we possess the therapeutic tools to treat depression successfully it remains one of the major tragedies that depression is still underdiagnosed and undertreated.
2. The Genetics Of Depression
After more than a century of family, twin, and adoption studies we can now state with certainty that almost all forms of behavior, including depressive psychopathology, are influenced by multiple genes. Such genes are called quantitative trait loci (QTLs) because they are likely to result in quantitative (continous) distributors of phenotypes that underlie a predisposition to depression. Specifically, this means that there is no single locus that by itself causes psychiatric syndromes: instead multiple alleles (gene variants) that interact to produce vulnerability are found at multiple loci within the genome. Genetic research in psychiatry has left little room for the supposition that a single gene at a given locus determines any given psychiatric syndrome. In other words, in different families, different gene variants are required to confer increased vulnerability. In the light of the manifold possibilities of gene–gene interaction, genetic research has provided a particularly interesting demonstration that genes do not act alone.
Genes must interact with nongenetic environmental factors to convert vulnerability into illness. This is an important aspect as much research in the past has been devoted to the understanding of the development of depression based entirely on an individual’s biography, making depression one of the last hermeneutic resorts in medicine. Many studies have shown that there is little reason to doubt that major stressful experiences, in particular those that occur in early life or even during pregnancy, render an individual more liable to develop depression (Heim et al. 2000). But it needs to be recognized that the conversion of life events into severe psychopathology requires the presence of the appropriate genetic endowment. An additional level of complexity has been introduced by the studies of Kenneth Kendler, who showed that individuals with a high genetic risk of depression are more likely to expose themselves to depression-triggering life events (Kendler et al. 1997). Indeed, several studies have supported this surprising proposition, that genetic factors may indirectly influence the liability to major depression by predisposing an individual to an aversive environment.
This shows that genetic and nongenetic environmental factors are much too inextricably intertwined to be dissected with high precision. Only when functional genomics have ultimately allowed us to establish a broad enough database on the functions of individual genes and gene–gene interactions, will the influence of environmental nongenetic factors on vulnerability to, and the onset and course of, depressive syndromes become available to analysis.
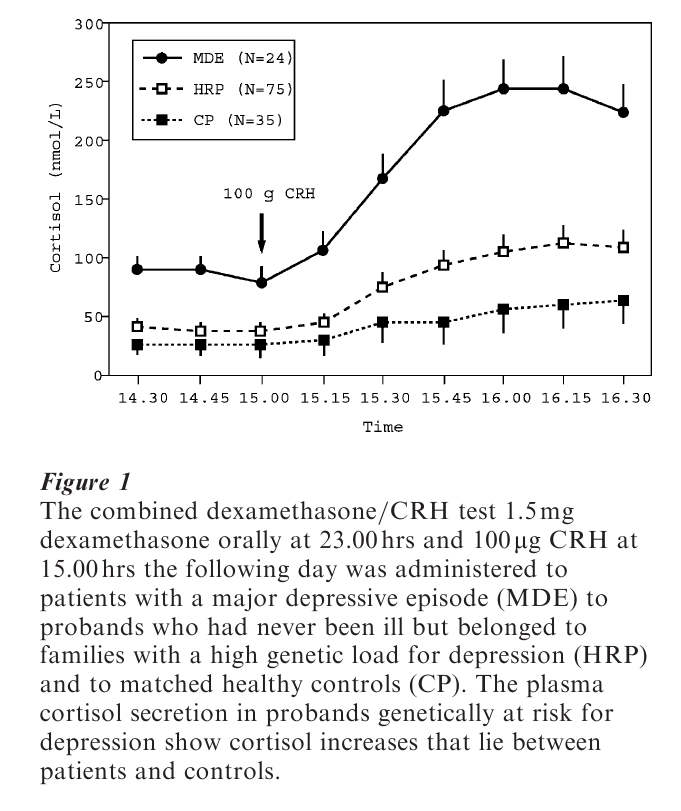
Definition of the depressive phenotype for genetic studies is extremely difficult and findings from these tudies have been largely inconclusive. Another reason for the paucity of genetic studies of unipolar major depression is the psychosocial adversity toward studies that call the stress diathesis into question. The use of association studies, which allow detection of small effects, have for methodological reasons (the need for the whole genome scan) been limited to candidate genes. The most intriguing of these studies is that of Klaus Peter Lesch, who investigated—together with colleagues from the National Institute of Mental Health, Bethesda, MD—the serotonin transporter gene, located on chromosome 17 (Lesch et al. 1996). This serotonin transporter is of particular interest as the majority of antidepressants act on this protein located in the cell membrane of presynaptic nerve terminals. By blocking the reuptake of serotonin molecules these drugs, of which Prozac is the best known, enhance serotonergic neurotransmission.
Lesch submits that a polymorphism in the transcriptional control region of the serotonin transporter may result in a long and a short version of the promoter polymorphism. The occurrence of the short allele has been shown to be associated not only with decreased transcriptional efficiency resulting in reduced serotonin transporter expression, but also with those personality traits that are likely to be relevant to vulnerability to depression. Most studies aimed at replicating this intriguing finding have failed to do so, but this does not invalidate the experimental approach. Rather, it suggests that the means we currently have to phenotype individuals are perhaps not suited to genetic studies. Researchers have questioned the usefulness of diagnostic algorithms such as the Diagnostic and Statistical Manual of Mental Disorders of the American Psychiatric Association (DSM-IV) or the International Classification of Disease of the World Health Organization (ICD-10) for studies of causality because the psychopathological symptoms that constitute psychiatric diagnoses may be the very remote effects of genes. Therefore, it has been suggested that it would be more fruitful if genetic studies focused on more direct measures of brain function.
Such a strategy is exemplified in the Munich Vulnerability Study conducted at the Max Planck Institute of Psychiatry, where individuals with no history of illness but, being members of families with a high genetic load for depression, are investigated not only according to psychopathology but also according to laboratory measurements and other symptoms that can be objectified. Among these laboratory assessments are neuroendocrine function tests and sleep electroencephalographic-activity recordings. As illustrated in Fig. 1, the hormonal response to a neuroendocrine function test specifically designed to identify impairments in stress hormone regulation is altered in probands with a high familial risk for depression. Their stress hormone release is significantly increased compared with matched controls. What is not yet known is to what extent premorbid stress hormone dysregulation may predict an increased likelihood of developing overt clinical depression.
The idea behind the strategy is to identify patients with impaired stress hormone regulation across diagnostic boundaries and to study their genetic commonalities. Of course, other symptoms or symptom clusters, e.g., psychotic symptoms, need to be considered regardless of the diagnostic category to which the individual patient may belong. The proposed breakdown of the diagnostic description of complex behavior into simpler components including specific symptoms, together with results from neurobiological investigations (e.g., functional brain imaging, electroencephalography, drug treatment response, neuroendocrine assessment, etc.) deserve more attention in future genetic research than do traditional diagnostic attributions.
Bipolar depression, where depressive episodes alternate with manic episodes characterized by elevated mood, ideas of grandiosity, flight of thought, sleeplessness, etc., is much more easily phenotyped. Attempts to understand this disorder in terms of biographic events have also not been very successful, thus facilitating a push towards genetic research. Morbid risk in first-degree relatives is four to six times higher than the population prevalence of 1% for both men and women, making the case for genetic susceptibility particularly obvious. Enormous efforts in the United States and Europe have allowed the study of families large enough to discover a linkage between bipolar disorder and certain areas on several chromosomes, notably 12q, 18p, 18q, 21q, 4p, and X1.
Of particular interest is a study by Nicholas Barden from Quebec, Canada, who completed a genome screen in a large French Canadian pedigree with over 100 individuals (Morissette et al. 1999). This investigator showed a linkage to chromosome 12q 23–24 in some, but not all branches of the pedigree and concluded from his data that more than one locus may be segregating in this isolated population. A smaller pedigree from the same geographic region also confirmed the linkage between bipolar depression and chromosome 12. Similar conclusions were drawn from another investigation into a family where major affective disorder was segregating with Darier’s disease. The gene, whose mutation for Darier’s disease encodes a calcium ion pump, was recently identified but screening for mutations in a sample of bipolar probands without Darier’s disease did not identify any gene mutation, rendering it unlikely that the susceptibility gene for bipolar disorder is in close proximity to the Darier’s disease gene.
This example illustrates that, despite promising results and a fairly clear phenotype (although it may well be that a bipolar case is diagnosed very late because a manic episode occasionally only becomes clinically apparent in later life), we are still far away from the identification of susceptibility genes for bipolar disorder. One conclusion to be drawn is that there are likely to be numerous susceptibility genes that influence the risk of unipolar or bipolar depression. Supported by progress in biotechnology, critical regions for each susceptibility gene will be narrowed down and candidate genes will be tested much faster than in the past. These opportunities, together with new techniques allowing the dissection of gene–gene and gene–environment interactions, will ultimately allow the identification of susceptibility genes. The future role of the clinician scientist will be to improve diagnosis-independent phenotyping and to conduct studies focused on the elucidation of potential pathophysiological roles of candidate genes.
3. Hypotheses On The Causality Of Depression At The End Of The Twentieth Century
3.1 Hormones and Neuropeptides
Depression is associated with a number of changes in hormonal secretion and conversely, certain endocrine disorders (i.e., hypothyroidism or Cushing’s disease) are associated with excessive psychiatric morbidity, most frequently depression. Because the peripheral hormonal readout is governed by hypothalamicreleasing factors, which are themselves subject to complex regulations involving biogenic amines (e.g., serotonin, norepinephrine) and amino acids (e.g., gammaaminobutyric acid, GABA, and N-methyl-Daspartat, NMDA) many researchers in the 1970s and 1980s had hoped that the neuroendocrine system might provide a ‘window to the brain.’ For example, growth hormone secretion from the pituitary was found to be blunted in patients with depression, following a variety of pharmacological interventions including the α-adrenoceptor system. Thus, the decreased growth hormone release following clonidine, an α-adrenoceptor agonist, was suggested as an indicator of desensitized adrenoceptor systems in these patients. Whereas the neuroendocrine approach that attempted to identify central neurotransmitter disturbances through measurements of hormonal responses to various pharmacological probes proved to be only of limited value in explaining the pathophysiology of depression, a new hypothesis emerged, linking stress hormone regulation to the development of depression and its course during drug treatment.
The fundament of this hypothesis is that elevated central neuropeptides accounting for increased peripheral stress hormone (corticotropin and cortisol) secretion in patients with depression, also mediate a number of psychopathological cardinal symptoms of depression. The neuropeptide in question is corticotropin-releasing hormone, CRH, that, when secreted from the hypothalamus, reaches the pituitary through portal vessels and elicits corticotropin from the pituitary and consequently cortisol from the adrenal cortex. CRH is also present in other brain areas, the frontal cortex, the amygdala, the hippocampus, and the locus coeruleus, all of which are implicated in the generation of stress-related psychopathology such as anxiety, cognitive function, sleep disturbance, appetite and sexual arousal, autonomous regulation and other features.
This has led to research activities from which clear evidence has emerged that CRH not only accounts for peripheral stress hormone secretion, but is also a key mediator of anxiety and depressive symptoms (Holsboer 1999). The question why CRH is elevated in some brain areas and not others is not yet clear, but some evidence has accumulated that a disturbed corticosteroid receptor function exists that prevents elevated cortisol levels from appropriately regulating CRH synthesis. Notably, the regulation of CRH gene expression by steroid activated corticosteroid receptors is very complex and varies across brain areas. The corticosteroid receptor hypothesis represents a paradigmatic shift inasmuch as that a therapeutic strategy has been developed from a clinical observation (Holsboer 2000). Blocking the action of CRH requires knowledge of the receptors through which this neuropeptide exerts its psychopathological effect. Molecular pharmacology and genetic research have helped us to identify the CRH receptor subtype (CRH1 receptor) responsible, and the discovery of small molecules that selectively antagonize CRH actions at this receptor have allowed us to test this CRH hypothesis.
Basic studies in animals and first clinical studies support the notion that CRH1 receptor antagonists are capable of improving signs and symptoms of stress-related clinical conditions, and that they are of potential value in treating major depression. This concept is frequently burdened by two serious misunderstandings that need to be addressed: the clinical efficacy of CRH receptor antagonists is not limited to patients showing peripheral hypercortisolemia. This endocrine symptom reflects hypothalamic–pituitary activity, a neuroanatomical axis, certainly not the primary locus of depressive pathology. CRH may be elevated in the amygdala, the hippocampus or the locus coeruleus, with or without concurrent elevation of hypothalamic CRH, peripheral corticotropin or cortisol. Second, hypersecretion of stress hormones does not reflect the emotional stress associated with depression. It is of note that many studies have shown a time-course pattern where an elevation of stress hormone release preceded the overt depressive syndrome. Moreover, a refined neuroendocrine test, the combined dexamethasone CRH test, has proved to be of value in identifying patients not showing depressive symptoms but being at risk of relapsing into a depressive episode within six months. This time pattern rejects the possibility that exaggerated stress hormone release is secondary to the emotional stress associated with depression.
3.2 Biogenic Amines
The traditional pathogenetic concepts of depression emerged from the discovery of the pharmacological properties of current antidepressants. Investigators in the United States led by Joe Schildkrauth (1965) formulated the catecholamine hypothesis of depression, submitting that depression is associated with a deficiency of catecholamine activity (particularly norepinephrine) at functionally important adrenoceptor sites in the brain, whereas mania is associated with a relatively excessive catecholaminergic neurotransmission. Norepinephrine-containing neurons project from the locus coeruleus, a brain stem nucleus, into almost all brain areas. Notably, CRH-containing nerve terminals innervate the locus coeruleus. Examples of altered norepinephrine activity are listed in Table 2.

A number of these findings are in agreement with a norepinephrine deficit. In particular, postmortem studies have shown that the locus coeruleus of depressed suicide victims contains increased levels of tyrosine hydroxylase, the rate-limiting enzyme of norepinephrine biosynthesis. This finding is in accordance with either premortem overactivity of adrenergic neurons or a deficit in the biosynthetic product norepinephrine. Similarly, in the same brain nucleus the levels of the presynaptic norepinephrine trans- porter are decreased, which can also be interpreted as a compensatory downregulation of this protein in response to decreased norepinephrine levels. However, what is puzzling is the failure to induce an aggravation of depressive symptomatology in depressed patients after treatment with a drug that inhibits norepinephrine biosynthesis.
The great acceptance of selective serotonin reuptake inhibitors (SSRIs), reflected by their major (over 70%) share of the total antidepressant market, has rekindled the interest in the serotonin hypothesis of depression originally submitted by Alec Coppen (1969) in the United Kingdom. This hypothesis proposed that decreased serotonergic activity accounts for the development and course of depression. These changes in serotonergic neurotransmission may be attributable to a decreased serotonin release, fewer serotonin receptors (of which exist more than 14 different subtypes), or impaired serotonin receptor-mediated signal transduction. Evidence for a serotonin deficiency, however, is conjectural and relies on (a) decreased 5-hydroxyindoleacetic acid (a principal metabolite of serotonin) concentrations in the cerebrospinal fluid, (b) a neuroendocrine function test using the pharmacological serotonin releaser fenfluramine, which induces blunted prolactin response in depressed patients, and (c) postmortem studies where indicators for serotonergic activity were found to be reduced (Delgado et al. 1994).
It is of note that serotonergic activity and stress hormone activity are closely intertwined. For example, if CRH is elevated in the hippocampus, the stress-elicited serotonin release is reduced and if CRH1 receptor capacity is reduced, either by targeted mutation of the CRH1 receptor or by CRH1 receptor antagonists, serotonin response to stress is elevated, suggesting that blocking CRH-mediated effects through CRH1 receptor antagonists restores impaired serotonergic activity.
4. Treatment Of Depression
The front line of therapy for severe major depression is drug treatment with antidepressants. The effect of these agents is assisted by psychotherapy that needs to be pragmatically adjusted to the patients’ needs. The antidepressant drugs available for treatment all work through modulating the neurotransmission of biogenic amines (see above). Some of them are fairly selective such as SSRIs (e.g., fluoxetine, paroxetine or citalopram), or norepinephrine reuptake inhibitors (e.g., reboxetine), while other drugs combine both pharmacologies (e.g., venlafaxine, nefazodone). Because of their pharmacological similarity, it is not surprising that only subtle differences in the clinical profiles of these drugs exist, and that antidepressants are consequently chosen by physicians according to their safety and tolerability profiles, provided their choice is not deterred by the higher costs in countries where reimbursement is limited.
While the superiority of antidepressants over placebo is undisputed among clinician scientists, the exact figures are still a matter of discussion (Quitkin et al. 2000). This is mainly related to the fact that most studies designed to provide the requisite statistical significance for monitoring side-effects and confirming efficacy use large outpatient samples that often contain only moderately depressed patients, have high dropout rates, and include the administration of suboptimal drug dosages. All these factors favor the placebo response, frequently found to be close to 50%, whereas the active drug often does not exceed a 75% response rate. Notably, response in these studies is defined as an improvement of 50% over baseline, which is clinically unsatisfactory and certainly not the desired treatment goal. Thus, the impression emerges from such studies that antidepressants improve the clinical condition by not more than 25%, which is unrealistic. A possible consequence of these figures comes from a study by the WHO which revealed that in patients who were diagnosed with depression, only 31.9% were prescribed an antidepressant, 25.5% received tranquilizers (benzodiazepines), and 19% received ‘miscellaneous’ drugs such as herbal remedies.
The latter includes St. John’s Wort, a drug that is prescribed to half the treated population in Germany, where psychotherapy is also much more widely available than in other countries. This reflects how diverse attitudes toward the pharmacological treatment of depression can be. A current study sponsored by the National Institute of Mental Health, will hopefully elucidate whether the widespread use of St. John’s Wort is scientifically justified.
The most important impediments in the pharmacotherapy of depression are (a) the lack of differential therapeutic aids, i.e., there is no tool available to select the appropriate drug for the individual patient; (b) the lack of objective laboratory signs and symptoms to monitor adequacy of treatment; (c) the protracted onset of action, which can take weeks or even months; and (d) side-effects, which are particularly disturbing and often lead to noncompliance after the patient has begun to respond, but still needs treatment to consolidate the beneficial therapeutic effect.
Some of these unmet needs no longer seem to be unsurmountable (Holsboer 2001): (a) differential therapy will be guided by genotyping of patients, helping the physicians to identify which drug is optimally suited, ultimately leading to customized therapies; (b) neuroendocrine function tests such as the combined dexamethasone CRH test have proved useful in providing feedback information about ongoing neurobiological pathology independently from acute psychopathology ratings. Its more widespread use holds the promise of shortening the course of treatment and hospitalization; (c) onset of action can possibly be shortened by enforced normalization of stress hormone excess via CRH receptor antagonists; and (d) side-effects can be partly reduced by customized drug therapies or by a shift to drugs that target neuropeptides instead of biogenic amine neurotransmitters. Neuropeptide hypersecretion primarily reflects a pathological neuronal hyperactivity, and therefore, neuropeptide receptor antagonists are effective only if neuropeptide hypersecretion exists.
5. Summary And Outlook
The lifetime risk of a major depression is high and, once suffering from depression, the morbidity for other clinical conditions, ranging from common infection to ischemic heart disease, escalates considerably. Thus, depression is already a major cause of disability and will become the most costly disease in the coming decades. In the light of this, any effort to improve the prevention and treatment of depression is justified. The evidence implicating genes as factors predisposing to depression is impressive but complex inheritance has undermined efforts so far to identify those susceptibility genes that interact with environmental factors to precipitate the disease. Despite the current excitement about the possibilities of genetics to identify ultimately a set of gene variants at many different loci that confer a predisposition to depression, I believe that the first revolutionary step will emerge from pharmacogenomics.
This research will elucidate inherited differences in sensitivity to drugs, allowing the physician to predict which particular drug would be best and which harmful for an individual patient. This possibility would be an enormous advantage over today’s trial and error strategy and, once feasible, would reduce the time before the right drug at the right dosage is administered. Another forthcoming major change that will not be unanimously embraced by all psychiatrists is the departure from diagnostic categories. These have been a stumbling block for psychiatric research in the past and will be even more so in a future that is dominated by functional genomics. Therefore, clinical research is challenged to phenotype patients more appropriately on psychopathological and neurobiological levels that cut across traditional diagnostic boundaries. The enormous potential of an interdisciplinary approach encompassing psychopathology, neurobiology, pharmacology, and genetics together, with progress in biotechnology, will lead to a body of knowledge so powerful that not only will better treatments become available, but ultimately, the causal factors underlying depression will also be exposed.
Bibliography:
- Coppen A J 1969 Biochemical aspects of depression. International Psychiatry Clinics 6: 53–81
- Delgado P L, Price L H, Miller H L, Salomon R M, Aghajanian G K, Heninger G R, Charney D S 1994 Serotonin and the neurobiology of depression: Effects of tryptophan depletion in drug-free depressed patients. Archives of General Psychiatry 51: 865–74
- Heim C, Newport D J, Heit S, Graham Y P, Wilcox M, Bonsall R, Miller A H, Nemeroff C B 2000 Pituitary–adrenal and autonomic responses to stress in women after sexual and physical abuse in childhood. Journal of the American Medical Association 284: 592–7
- Holsboer F 1999 The rationale for corticotropin-releasing hormone receptor CRH-R antagonists to treat depression and anxiety. Journal of Psychiatric Research 33: 181–214
- Holsboer F 2000 The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology 23: 477–501
- Holsboer F 2001 Prospects for antidepressant drug discovery. Biological Psychiatry, in press.
- Kendler K S, Karkowski-Shumann L 1997 Stressful life events and genetic liability to major depression genetic control of exposure to the environment? Psychological Medicine 27: 539–47
- Kessler R C, McGonagle K A, Zhao S, Nelson C B, Huges M, Eshleman S, Wittchen H U, Kendler K 1994 Lifetime and 12 month prevalence of DSM-III-R psychiatric disorder in the United States. Archives of General Psychiatry 51: 8–19
- Lesch K P, Bengel D, Heils A, Sabol S Z, Greenberg B D, Petri S, Benjamin J, Muller C R, Hamer D H, Murphy D L 1996 Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science 274: 1527–31
- Morissette J, Villeneuve A, Bordeleau L, Rochette D, Laberge C, Gagne B, Laprise C, Bouchard G, Plante M, Gobeil L, Shink E, Weissenbach J, Barden N 1999 Genome-wide search for linkage of bipolar affective disorders in a very large pedigree derived from a homogeneous population in Quebec points to a locus of major effect on chromosome 12q23-q24. American Journal of Medical Genetics 88: 567–87
- Nathan K I, Musselman D L, Schatzberg A F, Nemeroff C B 1995 Biology of mood disorders. In: Schatzberg A F, Nemeroff C B (eds.) The American Psychiatric Press Textbook of Psychopharmacology. American Psychiatric Press Inc, Washington, London, pp. 439–77
- Quitkin F M, Rabkin J G, Gerald J, Davis J M, Klein D F 2000 Validity of clinical trials of antidepressants. The American Journal of Psychiatry 157: 327–37
- Schildkraut J J 1965 The catecholamine hypothesis of affective disorders: A review of supporting evidence. The American Journal of Psychiatry 122: 509–22
ORDER HIGH QUALITY CUSTOM PAPER

Always on-time

Plagiarism-Free

100% Confidentiality