
View sample mental health research paper on biological and genetic determinants of mental health. Browse other research paper examples for more inspiration. If you need a thorough research paper written according to all the academic standards, you can always turn to our experienced writers for help. This is how your paper can get an A! Feel free to contact our writing service for professional assistance. We offer high-quality assignments for reasonable rates.
Introduction
The purpose of this research paper is to provide a broad overview of what we know about the genetic epidemiology of psychological and psychiatric ailments. As the title suggests, we provide a brief review of the genetic epidemiologic methods and principles that guide the search for genes influencing these conditions. We also evaluate the evidence implicating familial and genetic factors in general, and specific genes and alleles in particular. As one of the most heritable – and most often studied – psychiatric disorders, we will focus our review on schizophrenia; however, general principles will be emphasized, and particularly successful efforts directed at other mental disorders will also be highlighted where appropriate. This research paper concludes with a series of recommendations to hasten the discovery of risk genes for psychiatric disorders. Most psychiatric disorders exhibit complex, non-Mendelian inheritance patterns, and thus are thought to result from the joint effects of multiple genes and environmental factors. As such, we refer to the genes for psychiatric disorders as risk genes rather than disease genes, since none may be either necessary or sufficient to elicit illness.
Academic Writing, Editing, Proofreading, And Problem Solving Services
Get 10% OFF with 24START discount code
Behavioral Genetic Methods And Principles
Family Studies
The first question that must be asked and answered when attempting to delineate the genetic and environmental components of a complex psychiatric disorder is: Does the phenotype run in families?, or, Is this phenotype familial? This question can be answered through the use of family studies. The basic design of the family study begins with the ascertainment of a group of subjects who are affected by the disorder (cases) and a comparable group of control subjects who do not have the disorder. Next, the biological relatives of these index subjects, or probands, are ascertained and evaluated for the presence of the illness, or in some cases, subthreshold forms of the illness. The rate of the disorder among family members of affected probands is then compared to the rate of the disorder among family members of control probands to determine the familial risk or relative risk.
If a psychiatric disorder has a genetic etiology, then biological relatives of cases should have a higher likelihood than relatives of controls of carrying the gene or genes that influenced illness in their relative, and thus they should be at greater risk for the illness themselves. In addition, the risk to relatives of cases should be correlated with their degree of relationship to the proband, or the amount of genes they share in common. First-degree relatives such as parents, siblings, and children, share 50% of their genes, on average, with the proband. Thus, first-degree relatives of cases should be at greater risk for the disorder than second-degree relatives (grandparents, uncles, aunts, nephews, nieces, and half-siblings), who share only 25% of their genes with the proband.
Twin Studies
Once a disorder has been established as familial, it becomes necessary to determine if that pattern is attributable to the inheritance of genes or to shared familial and other environmental factors. It is also important to quantify the contribution that genes make relative to that made by environmental factors, as this may encourage or discourage future molecular genetic studies; it may also influence the decisions made by individuals seeking genetic counseling and the usefulness of family information in identifying high-risk individuals to target for early intervention and prevention protocols. These questions can be answered by both twin studies and adoption studies but, mostly because of their relative ease to conduct and thus, their popularity, we focus on twin studies in this research paper.
In twin study designs, identical (monozygotic (MZ)), and fraternal (dizygotic (DZ)) twin-pairs are ascertained if at least one member of the pair is affected with a disorder. Twin-pairs are deemed concordant if both members of the pair have the illness, and are deemed discordant if only one member of the pair is affected. The ratio of concordant: discordant MZ twin-pairs is then compared to the ratio of concordant: discordant DZ twin-pairs.
MZ twins are derived from the same zygote and thus share 100% of their genetic material. In contrast, DZ twins result from separate fertilizations and thus share, on average, 50% of their genes – no more or less than any other pair of siblings. Thus, a typical MZ twinpair will have 50% more genes in common than a typical DZ twin-pair. However, the degree of similarity in environmental exposures between members of a MZ twinpair should be no different than that between members of a DZ twin-pair. Thus, any difference in concordance for a mental illness between the two types of twin-pairs can be attributed to the effects of the additional gene-sharing in the MZ twins.
If the concordance for a mental illness is higher among MZ twin-pairs than DZ twin-pairs, this is a good indication that there is a genetic contribution to the disorder; if MZ and DZ twin-pairs have approximately equal concordance rates, environmental factors are more strongly implicated. Frequently, concordance rates among twin-pairs are used to estimate the heritability of a disorder. Heritability measures the degree to which genetic factors influence variability in the manifestation of the phenotype. Heritability in the broad sense is the ratio of genetic to phenotypic variances, or the proportion of variance in schizophrenia risk that is accounted for by variability in genetic factors. A heritability of 1.0 indicates that all variability in the phenotype is due to genetic factors alone. In contrast, a heritability of zero attributes all phenotypic variation to environmental factors. Two points need to be carefully considered when interpreting heritability estimates, however. The first is that the heritability estimate is a maximum value, but the true heritability may be lower than its estimate in any given sample. The second is that heritability estimates are often context-dependent, and this is reflected by the fact that the heritability estimate accounts for the main effects of genetic factors but also gene-by-environment interactions. Thus, the entire heritability of a trait may be accounted for by the presence or absence of a necessary environmental cofactor. This is easiest to understand in the context of nonpsychiatric disorders, such as lung cancer. In the absence of important environmental cofactors such as exposure to cigarette smoke, the heritability would be much lower; in the case of phenylketonuria, lack of exposure to phenylalanine would reduce the heritability estimate to zero, since the condition would not occur at all. Recently, evidence of gene-by-environment interactions in psychiatry have been discovered and, in some cases, replicated. One such example of this is a commonly observed interaction of stress and a polymorphism in the serotonin transporter gene, which combine to increase the risk for major depression beyond the main effect of either risk factor.
General Familial And Genetic Factors Implicated In Mental Disorders
Family Studies
As shown in Table 1, many disorders identified in the Diagnostic and Statistical Manual of Mental Disorder, Fourth Edition (DSM-IV) (American Psychiatric Association, 1994) have been evaluated within the framework of a family study and have yielded an estimate of the risk of illness to a sibling of an affected person (ls). For the most part, familial risk estimates for psychiatric conditions have been found to range from 4 to 12, indicating that, depending on the disorder, a sibling of an affected individual is four to 12 times more likely to develop the disorder himself as compared to an individual from the general population.
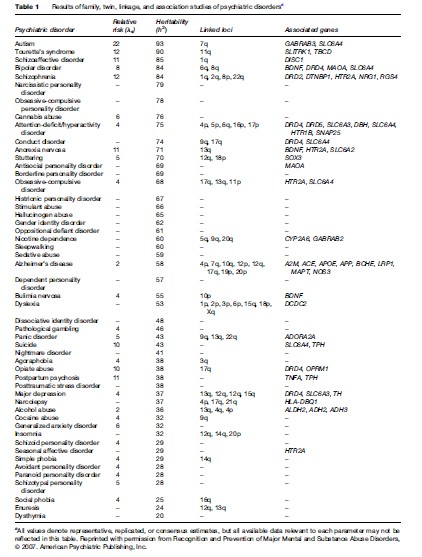
It is noteworthy, however, that most family studies of psychiatric disorders that have included second and third-degree relatives of the proband have observed a nonlinear relationship between relative risk for illness and degree of relationship to the proband (i.e., expected proportion of shared genes). Thus, for example, the consensus from studies of schizophrenia is that second-degree relatives of schizophrenia patients are at an approximately two to threefold increased risk of illness, but first-degree relatives are at a tenfold increased risk despite a mere doubling of expected alleles shared with the proband. Furthermore, the risk to offspring of two schizophrenic parents, from whom the affected offspring received all of their genes, is not absolute ( 46%); this pattern is also observed in studies of most other psychiatric disorders. These results underscore the complexity of the genetic bases for psychiatric disorders and imply that gene–gene interactions (epistasis) as well as environmental factors must contribute to their etiologies.
Collectively, the evidence from family studies suggests that psychiatric conditions do aggregate in families, that multiple genes and environmental factors may be involved in these illnesses, and that these disorders can present (even within families) with different degrees of severity. Despite this powerful evidence, it is important to recognize that familiality does not necessarily establish heritability. For example, religion and language are familial traits, as all members of the same family often practice the same religion and speak the same language. These facts do not reflect the transmission of religion genes or language genes through the family, but rather the common environment and upbringing that those family members share.
Twin Studies
The suggestions raised by these family studies can be formally evaluated through the use of twin studies, and as can be seen in Table 1, the heritability of many psychiatric disorders has already been examined. Of those psychiatric disorders described in the DSM-IV that have been the subject of a twin study, all but one was found to be significantly heritable. Estimates of the heritability of these disorders vary widely, from a high of more than 90% for autism (Bailey et al., 1995) to a low of 0% for dysthymia (Lyons et al., 1998). Thus, while some psychiatric disorders may have no genetic basis, most have a genetic component and several are predominantly attributable to genes.
Analogous to the results of family studies, MZ twins often have a rate of concordance for a psychiatric illness that is greater than twice that observed among DZ twins, even though their expected proportion of shared genes is only double that of DZ twins. For example, the best evidence from twin studies of schizophrenia suggests a rate of concordance of approximately 46–53% for MZ twins and 14–15% for DZ twins. These data further support the possibility of epistasis in the etiology of the disorder, and similar results have been observed in twin studies of other psychiatric conditions. Furthermore, MZ twins are not 100% concordant for any psychiatric disorder, confirming that environmental factors make a strong contribution to the overall risk, even for the most heritable mental disorders.
Molecular Genetic Methods And Principles
Linkage Studies
Knowing that genetic factors are involved in the etiology of a psychiatric disorder – and to what degree – is essential for designing optimal molecular genetic studies to reveal the chromosomal location of the responsible risk genes. To identify regions of chromosomes that have a high likelihood of harboring risk genes for an illness, linkage analysis is a highly appropriate strategy. Families are ascertained for linkage analysis through a proband affected with the disorder of interest. Each individual in the family is then genotyped at a series of DNA markers (not necessarily in genes) spaced evenly throughout the genome, and the cosegregation of these DNA markers with the illness is tracked in each pedigree. Evidence for cosegregation at each marker locus is summed across pedigrees to derive an index of the likelihood of the obtained patterns of marker-phenotype cosegregation given the sampled pedigree structures.
Although the DNA markers used for linkage analysis are not presumed to be actual risk genes for the disorder, they are numerous and dense enough to ensure that their coinheritance with a nearby (but unobserved) risk gene could be inferred with reasonable certainty based on the coinheritance of the marker with the phenotype that is influenced by that risk gene. In this design, the disorder serves as a proxy for the risk gene; thus DNA markers that cosegregate commonly with the disorder are presumed to cosegregate commonly with its underlying risk gene. Because the probability of cosegregation of two pieces of DNA is inversely proportional to the distance between them, the regularity of the cosegregation of the DNA marker and the disorder gives an indirect indication of the genetic distance between the DNA marker and the unobserved risk gene.
The possible outcomes of a linkage analysis will vary based on the structure of families ascertained for analysis. For example, linkage analysis can be performed with affected sibling-pairs, or with other affected relativepairs, or with small nuclear families, or with large extended pedigrees. Regardless of what family structure is the principal unit of analysis, the common output across methods is some index of the degree of phenotypic similarity of family members and the degree of genotypic similarity between those individuals at each DNA marker. These indices are summed across families to determine the overall evidence for linkage at a given locus in the full sample. If a given DNA marker cosegregates with illness through families more often than would be expected by chance, this indicates that the marker is linked (i.e., is in relatively close physical proximity) to a risk gene that influences expression of the disorder.
Association Studies
Once regions of certain chromosomes have been implicated from linkage analysis as harboring a risk gene for a disorder, the next step is to identify what specific gene is segregating through families to give rise to that linkage signal. A gene can be selected for such analysis subsequent to linkage analysis as a means to follow up on evidence for increased genetic similarity at a locus among affected individuals in a family (i.e., a positional candidate gene approach). Alternatively, specific genes can be examined in the absence of linkage information if there is some compelling physiological reason to suspect that the gene influences risk for a given disorder (i.e., a functional candidate gene approach). For example, dopamine-system genes, such as receptors and transporters, are commonly examined as functional candidates for schizophrenia, mood disorders, substance use disorders, and ADHD. In contrast to linkage analysis, which uses random DNA markers as proxies for nearby risk genes, genetic association analysis is an appropriate method for determining if a particular gene variant has a direct effect on risk for an illness, or is in tight linkage disequilibrium with such a gene. Recent developments in whole-genome association scan methods will marry the unbiased genomic survey approach of linkage analysis with the power and precision of association analysis and may shed new insights on previously unsuspected candidate genes for psychiatric disorders.
If a gene influences risk for a mental illness, this should be detectable as an increased frequency of the risk allele of the gene in cases compared to controls. Within the context of the family, this would be detectable as an increased likelihood of an affected patient receiving the risk allele of the gene from his parent, even when both the risk and normal forms of the gene were present in the parent and should have been transmitted to offspring with equal frequency and likelihood.
Specific Chromosomal Loci And Genes Implicated In Psychiatric Disorders
Linkage Studies
For the most heritable psychiatric disorders, including schizophrenia, bipolar disorder, and autism, numerous independent genome-wide linkage analyses have been performed (Table 1). In fact, each of these three disorders has been studied often enough by linkage analysis to allow for the quantitative combination of evidence across studies by meta-analysis. For autism, two meta-analyses have confirmed linkage to chromosome 7q, which was observed in several individual studies (Badner and Gershon, 2002a, 2002b; Trikalinos et al. 2006). Schizophrenia and bipolar disorder have also been subjected to more than one independent meta-analysis each, but with less agreement between the methods than has been observed for autism.
For schizophrenia, no less than 18 independent genome-wide linkage analyses have been published to date. Each of these studies has identified at least one chromosomal region in which either significant or suggestive evidence for linkage was observed. Unfortunately, the major findings from these genome-wide linkage scans do not, on first glance, appear to overlap to any great extent. Badner and Gershon (2002a) performed the first meta-analysis of these genome-wide linkage scans, and the results of their pooled analysis identified loci on chromosomes 8p, 13q, and 22q as the best candidates for harboring schizophrenia risk genes. Other promising regions included 1q, 2q, 6q, and 15q, but evidence for linkage at these loci was weaker, indicating a need for further replication. Subsequently, Lewis et al. (2003) conducted a meta-analysis of these and additional studies using an alternate methodology. Their results were somewhat different, identifying chromosome 2q as the prime candidate linked locus, and revealing somewhat weaker evidence for linkage on chromosomes 1q, 3p, 5q, 6p, 8p, 11q, 14p, 20q, and 22q. However, both meta-analyses were consistent in identifying chromosomes 1q, 2q, 8p, and 22q as the most reliably linked loci across individual studies.
For bipolar disorder, the first meta-analysis (that of Badner and Gershon, 2002a) found the strongest evidence for significant linkage on chromosomes 13q and 22q. (Of note, these were two of the three loci these authors also identified as linked to schizophrenia.) In stark contrast, the meta-analysis of Segurado et al. (2003) found the strongest evidence for linkage at loci on chromosomes 9p, 10q, and 14q. Most recently, a combined analysis of primary genotype data (rather than pooled study-level results) from all 11 studies implicated chromosomes 6q and 8q as the strongest candidates for harboring risk genes for bipolar disorder, perhaps providing the best evidence to date on the topic.
Genome-wide linkage analyses have thus provided some strong leads (but some ambiguous ones as well) in the search for loci harboring risk genes for some mental illnesses; however, the method is certainly not optimal for detecting genes with small effects on risk. For example, Risch and Merikangas (1996) illustrated that a locus conferring four times greater risk for a disorder could be detected by linkage analysis in 200–4000 families; a reasonable number for today’s large, multisite collaborative research studies. However, to detect a locus that increases risk by only 50%, a minimum of 18 000 families would be needed. This number of families is clearly unattainable by any single research group and, indeed, is beyond the reach of even the most effective research consortia. In fact, this number exceeds the total number of families studied to date in all published linkage analyses of schizophrenia, bipolar disorder, and autism combined. Therefore, for all practical purposes, we have reached an era where linkage analysis may no longer be a feasible strategy to detect genes that have a small but reliable influence on risk for complex mental disorders, especially those disorders with less of a heritable component to their etiologies.
Association Studies
When reviewing the status of behavioral genetic studies of psychiatric disorders, we saw that fewer conditions had been subjected to family studies than to twin studies, even though the former have traditionally preceded the latter in the chain of psychiatric genetic research (Faraone et al., 1999). This may be due to the ease of ascertaining twins rather than entire families, as well as the enhanced inferential power afforded by the twin study method. Thus, while family studies give an indication of the familial aggregation of a disorder (which may or may not reflect genetic factors), a twin study can directly establish whether or not genes influence the disorder and to what degree. This same type of reversal is also true for molecular genetic studies, wherein linkage studies have recently become far outnumbered by association studies. Again, this may be due to the ease of ascertaining units of analysis for an association study (i.e., unrelated cases and controls or, at most, small nuclear families with one affected individual) relative to a linkage study (i.e., affected sibling-pairs or extended pedigrees). In addition, the information gleaned from an association study may be more direct than that from a linkage study, since the former can test for direct effects on risk for each studied polymorphism, while the latter only identifies linked loci that must then be subjected to further finemapping to identify risk-conferring genes. Furthermore, in contrast to linkage analysis, association analysis should be more effective at detecting genes with small effects on liability. For example, Risch and Merikangas (1996) showed that a locus increasing risk for a complex disorder by 50% could be detected in as few as 950 subjects. This number of samples is more feasible than that needed to detect the same effect by linkage analysis (i.e., 18 000 families). In fact, many of the pooled association studies conducted to date have attained such numbers of subjects.
Due to these favorable attributes of association methods, many studies (we estimate over 2700) of functional and positional candidate genes for psychiatric disorders have been conducted over the past two decades; however, only a handful of genes exhibit reasonably strong evidence for exerting reliable risk for one or more of these disorders (Table 1). The genes listed here are those that have been implicated by (1) strongly significant evidence from a very large primary study; (2) significant evidence from two or more independent research groups; or (3) significant pooled evidence from meta-analysis. Although the genes listed in this table represent relatively strong candidates for these disorders, we must reiterate at this point that none of these genes are proven risk factors for an illness, and none is either necessary or sufficient for producing any psychiatric disorder. In fact, most have been found to increase risk less than twofold and account for only a small portion of the aggregate risk for the given mental illness in the population. In addition, the risk alleles and haplotypes identified in one sample are often not the same as those implicated in other studies. Thus, more work is needed to definitively specify the nature and magnitude of the influences of these genes on risk for the various mental disorders to which they have been associated.
Despite these successes, association studies remain plagued by some limitations, including their propensity for producing false-positive results (Lohmueller et al., 2003) and their limited breadth. Regarding the former, genes identified as associated with a mental illness in an initial study often overestimate the true effect size and subsequently fall victim to the winner’s curse, wherein the same magnitude of an effect cannot be replicated (Ioannidis et al., 2001; Glatt et al. 2003). As such, independent replication of genetic associations must be considered crucial for determining the role of a gene in a mental disorder. Regarding the latter, association methods have traditionally focused on one or at most a handful of genes at once, whereas linkage analysis constitutes a genome-wide survey of (relatively) unselected markers. Unfortunately, the prior probability of selecting the right candidate gene (out of 25 000 human genes) and the right polymorphism (out of more than 10 000 000 in the human genome) for analysis is remote. Most candidate genes for mental disorders have been targeted based on their expression within systems widely implicated in the disorder (e.g., functional candidate genes in the dopamine neurotransmitter systems in schizophrenia). This approach has thus far proven essential for clarifying the nature of dysfunction within these recognized candidate pathways; however, it may not be optimal for identifying additional novel risk factors outside of these systems. The recent advancement of laboratory and statistical methods for genome-wide association analysis should allow for a more unbiased examination of association patterns throughout the genome and help resolve this dilemma in coming years (Thomas et al., 2005).
Recommendations For Future Research
It is clear that the multifactorial polygenic etiologies and heterogeneity of psychiatric disorders obscure the discovery of their underlying genetic bases. The multifactorial polygenic nature of these diseases dictates that the signals obtained in linkage and association studies will be numerous and of very low intensity, while the heterogeneity of these disorders increases the noise against which these already-faint signals must be detected. Some of the most effective ways of combating these complexities are through the maximization of power, both by increasing sample size and by studying homogeneous groups of affected individuals. To identify the numerous genes with small effects on risk for psychiatric illness, various data-pooling strategies, such as direct combination of primary data or combination of study-level data by meta-analysis, can be particularly effective. However, to overcome the obstacles introduced by heterogeneity, delineating and studying smaller, more homogeneous subgroups of affected individuals may have the greatest beneficial effects on power. When used in tandem, the practices of pooling and splitting will yield maximal power for genetic studies, and hasten the discovery of the full compendium of risk genes for each heritable psychiatric condition. Other promising avenues for elucidating risk genes include the analysis of gene–gene and gene–environment interactions. An often overlooked potential benefit of genetic research on mental disorders is that, once risk genes are identified, environmental risk factors may become easier to detect within subgroups of subjects who do or do not possess those risk genes.
Since the first draft of the human genome was produced in 2001 (Lander et al., 2001), scientists have been touting the promise of genetic analysis for identifying risk factors for common diseases and the development of personalized medicine. Yet, the vast potential of genetic studies to change the clinical practice of psychiatry remains almost entirely untapped. Presently, the results of behavior genetic analyses, such as the family and twin studies described in this research paper, can be useful in genetic counseling situations to inform individuals of their chances of becoming affected with a particular illness. In addition, this information can be used prospectively for family planning and to help parents understand the risks for mental illnesses that may be carried by their children. However, families still cannot be examined for linkage at a particular locus to determine if they exhibit a particular pattern of marker transmission that suggests who in the family is at risk. Similarly, individuals in the population cannot be tested for the possession of particular genotypes to determine their cumulative risk of developing a particular disorder. The leads that have been generated from molecular genetic studies are simply not yet understood well enough to allow such uses. However, with continued advancement of laboratory and analytic methods, reliable risk genes for some psychiatric disorders are sure to emerge in the coming years.
Once genetic risk factors for psychiatric conditions become established and widely recognized, they can serve many purposes for early intervention and prevention efforts. For example, an objective, gene-based laboratory test could facilitate the arrival at a primary or differential diagnosis much more quickly than is presently possible. This in turn could speed the initiation of appropriate treatments, which consequently may promote better prognoses (McGlashan, 1999). Ultimately, a panel of genetic markers for a mental illness might be administered to high-risk individuals from affected families or in the general population to determine their likelihood of progression toward illness even before any clinical symptoms are manifest, which would allow these individuals to be targeted for early intervention and prevention efforts as well. Ultimately, this line of investigation may foster medicinal chemistry applications and the development of novel therapeutics that more precisely target faulty DNA sequences in aberrant genes or active sites in its protein. While recent successes in identifying specific risk genes for mental disorders are encouraging, much more work is needed to replicate and refine these results, and translate them into meaningful clinical applications.
Bibliography:
- American Psychiatric Association (1994) Diagnostic and Statistical Manual of Mental Disorders (DSM-IV). Washington, DC: American Psychiatric Association.
- Badner JA and Gershon ES (2002a) Meta-analysis of whole-genome linkage scans of bipolar disorder and schizophrenia. Molecular Psychiatry 7: 405–411.
- Badner JA and Gershon ES (2002b) Regional meta-analysis of published data supports linkage of autism with markers on chromosome 7. Molecular Psychiatry 7: 56–66.
- Bailey A, LeCouteur A, Gottesman I, et al. (1995) Autism as a strongly genetic disorder: Evidence from a British twin study. Psychological Medicine 25: 63–77.
- Faraone SV, Tsuang D, and Tsuang MT (1999) Genetics of Mental Disorders: A Guide for Students, Clinicians, and Researchers. New York: Guilford.
- Glatt SJ, Faraone SV, and Tsuang MT (2003) Meta-analysis identifies an association between the dopamine D2 receptor gene and schizophrenia. Molecular Psychiatry 8: 911–915.
- Ioannidis JP, Ntzani EE, Trikalinos TA, and Contopoulos-Ioannidis DG (2001) Replication validity of genetic association studies. Nature Genetics 29: 306–309.
- Lander ES, Linton LM, Birren B, et al. (2001) Initial sequencing and analysis of the human genome. Nature 409: 860–921.
- Lewis CM, Levinson DF, Wise LH, et al. (2003) Genome scan meta-analysis of schizophrenia and bipolar disorder, part II: Schizophrenia. American Journal of Human Genetics 73: 34–48.
- Lohmueller KE, Pearce CL, Pike M, Lander ES, and Hirschhorn JN (2003) Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nature Genetics 33: 177–182.
- Lyons MJ, Eisen SA, Goldberg J, et al. (1998) A registry based twin study of depression in men. Archives of General Psychiatry 55: 468–472.
- McGlashan TH (1999) Duration of untreated psychosis in first-episode schizophrenia: Marker or determinant of course? Biological Psychiatry 46: 899–907.
- Risch N and Merikangas K (1996) The future of genetic studies of complex human diseases. Science 273: 1516–1517.
- Segurado R, Detera-Wadleigh SD, Levinson DF, et al. (2003) Genome scan meta-analysis of schizophrenia and bipolar disorder, part III: Bipolar disorder. American Journal of Human Genetics 73: 49–62.
- Thomas DC, Haile RW, and Duggan D (2005) Recent developments in genomewide association scans: A workshop summary and review. American Journal of Human Genetics 77: 337–345.
- Trikalinos TA, Karvouni A, Zintzaras E, et al. (2006) A heterogeneity-based genome search meta-analysis for autismspectrum disorders. Molecular Psychiatry 11: 29–36.
- McGuffin P, Owen MJ, and Gottesman II (2002) Psychiatric Genetics and Genomics. New York: Oxford University Press.
ORDER HIGH QUALITY CUSTOM PAPER

Always on-time

Plagiarism-Free

100% Confidentiality