
Sample Dementia Research Paper. Browse other research paper examples and check the list of research paper topics for more inspiration. iResearchNet offers academic assignment help for students all over the world: writing from scratch, editing, proofreading, problem solving, from essays to dissertations, from humanities to STEM. We offer full confidentiality, safe payment, originality, and money-back guarantee. Secure your academic success with our risk-free services.
Dementia is a medical condition which is characterized by a generalized mental deterioration. The word has Latin roots, ‘de’ meaning separation, cessation or contraction, and ‘meme’ meaning ‘mind.’ Therefore, in dementia there is a cessation or contraction of the mind.
Academic Writing, Editing, Proofreading, And Problem Solving Services
Get 10% OFF with 24START discount code
The usage of the term dementia can be traced back to the Roman writer and encyclopedist, Aulus Cornelius Celsus in the first century AD. Celsus distinguished dementia, a chronic ‘contraction of the mind,’ from acute conditions such as delirium. The first medical identification of aging with dementia can be traced to Aretaeus of Cappadocia in the second century AD. He distinguished a condition which began in old age and continued until death, characterized by ‘a torpor of the senses, and a stupefaction of the gnostic and intellectual faculties’ (Adams 1861). Galen, perhaps the most influential physician of all time, also identified old age as a cause of dementia in the second century AD. He defined ‘morosis’ as a condition in which ‘the knowledge of letters and other acts are totally obliterated, indeed they can’t even remember their own names’ (Galen 1821–33 translation). One condition associated with morosis was old age.
No further advances in the clinical description of dementia appear to have been made for more than 1500 years. In 1793, the eminent American physician, Benjamin Rush, a signer of the United States Declaration of Independence and author of the first American textbook of psychiatry, a man who is credited by the American Psychiatric Association with being the father of American psychiatry, added to the clinical description of dementia. He noted that:
it would be sufficiently humbling to human nature if our bodies exhibited in old age the marks only of a second childhood, but human weakness descends even lower. I met with an instance of a woman between 80 and 90 who exhibited the marks of a second infancy, by such a total loss of her mental faculties as to lose all consciousness in discharging her alvine and urinary excretions. In this state of the body, a disposition to sleep succeeds the wakefulness of the first stages of old age’ (Rush 1793).
The accuracy of Benjamin Rush’s description has become increasingly apparent over the past two centuries.
The modern term, ‘senile dementia,’ is derived from Esquirol (1838), who described this condition in a French textbook of psychiatry. He noted that this is a condition in which there occurs a weakening of the memory for recent experience and a loss of drive and will power. He noted that senile dementia (in French, demence senile), appears gradually and may be ac-companied by emotional disturbances.
In modern usage, the term dementia refers to either: (a) any condition associated with a generalized mental deterioration, or (b) a progressive generalized mental deterioration, frequently occurring in later life. Both usages are commonly applied.
1. Progressive Dementia
The most important progressive dementia is Alzheimer’s disease, which plays a role in a majority of cases (Tomlinson et al. 1970). Other progressive dementias include cerebrovascular dementia, Lewy body dementia, and the frontotemporal dementias. With the exception of the frontotemporal dementias, these conditions increase in occurrence with increasing age. Consequently, the modern increase in lifespan in most regions throughout the world has been accompanied by a dramatic increase in the occurrence of these conditions (Henderson and Jorm 2000).
1.1 Alzheimer’s Disease
Alzheimer’s disease (AD) is one of the most devastating medical conditions of contemporary times. For example, in the United States, the number of persons in nursing homes and related institutions with AD exceeds the total number of persons in all hospitals and related institutions. Consequently, the institutional burden of AD in the United States is approximately the same as the institutional burden of all other illnesses combined. Worldwide, AD is estimated to afflict more than 15 million persons, and the prevalence continues to increase with increasing life expectancy of the world’s people.

The precise mechanism of the cause of AD is unknown. However, particular genotypes which pre-dispose to early onset AD have been identified. These genetic ‘defects’ are β-amyloid precursor protein mutations located on chromosome 21, presenilin 1 mutations located on chromosome 14, and presenilin 2 mutations which are located on chromosome 1. Some investigators point out that each of these mutations is associated with increased production of β-amyloid protein, one of the neuropathologic hallmarks of AD (Selkoe 1997). However, the presenilin mutations in particular, have been related to a variety of other pathologies apart from increased β-amyloid protein. Further evidence for the role of β-amyloid in the etiopathogenesis of AD is the observation that persons with Down’s syndrome develop Alzheimer’s path-ology early in life and carry an extra copy of the β-amyloid precursor protein gene resulting in excess β-amyloid production.

The occurrence of the common late-life form of AD has also been associated with particular genotypes. The most clearly identified gene which influences the occurrence of late life, also known as ‘sporadic’ Alzheimer’s disease, is the apolipoprotein E (apoE) gene (National Institute on Aging Alzheimer’s Association Working Group 1996). Caucasians who carry the apoE4 allele are at increased risk for AD in later life. Caucasians who are homogenous for apoE4 have as much as eight times the risk of AD as similarly aged persons who do not carry the apoE4 allele. Other genes and gene interactions which influence the occurrence of the common, late-life form of AD have not yet been fully elucidated.
Although the mechanism by which AD occurs is not known, a characteristic clinical course of AD has been described (Reisberg et al. 1982, 2000). This characteristic clinical course is accompanied by characteristic neuropathologic manifestations and a characteristic neuropathologic progression.
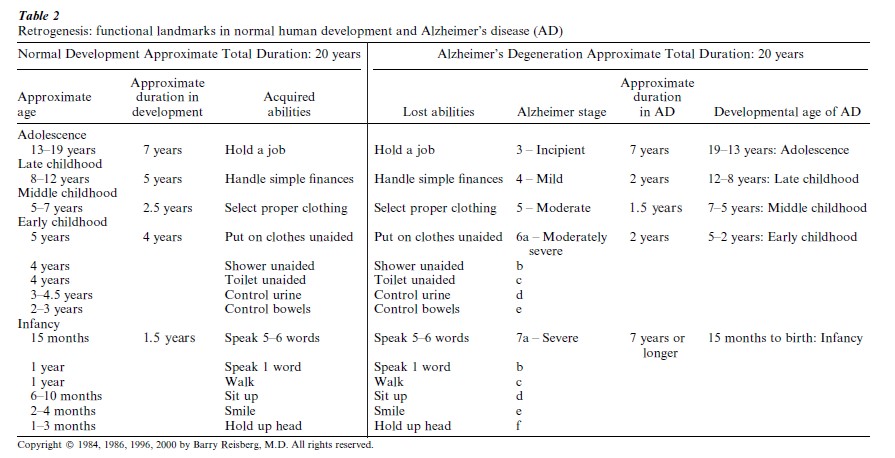
Clinically, AD can be described on a continuum with the changes in what is now termed normal aging, progressing to mild cognitive impairment (MCI) and subsequently to AD. This clinical continuum is illustrated in terms of the cognitive, functional and emotional progression of aging and AD in Table 1. Functionally, the progression of brain aging, MCI and AD can be described in even greater detail, in terms which are universally clear to both lay persons and professionals. This characteristic functional progression is familiar to many observers because it reverses the sequence of functional acquisition in human development, a phenomenon which has been termed ‘retrogenesis’ (Table 2) (Reisberg et al. 1999, 2000) The hallmark neuropathologic manifestations of AD are extracellular senile plaques which contain the β-amyloid protein, and intracellular neurofibrillary tangles which contain the protein tau. Very strong relationships have been found between the clinical progression of AD and the progression of pathology in the hippo-campus, a brain region which has been notably associated with memory. Progressive pathologic manifestations in the hippocampus include neurofibrillary pathology, volume loss, and neuronal cellular loss. Although the hippocampus is a brain region which manifests early and progressive AD pathology, AD is a generalized brain disease with, for example, progressive decrements in brain glucose utilization (energy metabolism) and progressive slowing of brain wave electrical activity on the electroencephalogram, as well as neuropathologic manifestations in numerous brain regions, which become particularly evident as AD progresses.
Neuropathologic stages of AD have been described (Braak and Braak 1991). These are transentorhinal stages I and II, limbic stages III and IV, and neo-cortical stages V and VI. Each of these stages of Braak and Braak are based upon the evolution and spread of neurofibrillary pathology in the brain of the AD patient.
Braak and Braak (1996) and Reisberg et al. (1992, 1999) have described a common basis for the neuropathologic and clinical manifestations of AD. This is the progressive involvement of the most recently, and therefore the most thinly, myelinated brain regions, followed by progressively more thickly myelinated, and therefore, better protected, brain regions. The most thickly myelinated brain regions subsume the functions and associated cognitive skills, which are the first to be acquired in the course of human development. Conversely, the most thinly myelinated brain regions subsume the most recently acquired functions and associated cognition. These recently myelinated regions are the most vulnerable to AD pathology. Consequently, cognitive and functional skills which are the most recently acquired are the most vulnerable to AD pathology, and those cognitive and functional skills which are the first to be acquired in normal human development are the last to be lost with the progression of AD. In brief, last in, first out. This retrogenesis phenomenon, which is illustrated for functional skills in Table 2, also applies to cognition and many aspects of brain physiology, including neurologic reflexes and apparently, to at least some extent, brain electrophysiology and metabolism.
Very recently it has been demonstrated that the β-amyloid, the major putative toxic pathology in AD is destructive to the oligodendroglia, the brain cells which produce the myelin (Xu et al. 2001). ApoE, the AD risk-determining factor, is a molecule associated with cholesterol transport. ApoE has been shown to affect neuronal repair mechanisms and the apoE3 allele appears to have a greater neuroprotective effect than the apoE4 allele (Poirier et al. 1993; Buttini et al. 1999). Therefore, a common basis for the molecular, pathologic, and clinical manifestations of AD can be postulated although many elements of the precise pathogenic mechanism remain unknown.
1.2 Vascular Dementia
Formerly, this entity was designated multi-infarct dementia because of its strong relationship to strokes, both large and small (Hachinski et al. 1974). Although originally considered by Hachinski to be a stroke-like entity with an abrupt onset and stepwise pattern of deterioration, this deterioration pattern is rarely observed. Vascular dementia is now thought to be a condition marked by progressive decline in cognition and functioning in which cerebrovascular, i.e., stroke risk factors, play a major role. These risk factors include hypertension and cardiovascular disease, as well as more overt evidence of cerebrovascular disease such as transientischemic attacks and focal neurologic signs and symptoms. In most cases, vascular dementia occurs in association with Alzheimer’s brain path-ology, a condition which is sometimes termed ‘mixed dementia.’ Much less frequently, vascular dementia appears to occur without evidence of concurrent AD. Consequently, many investigators view vascular dementia as on a continuum with AD. This continuum concept has become particularly compelling in recent years, since virtually all of the elements associated with vascular dementia have been shown to be risk factors for so-called ‘pure AD.’
In accordance with the concept of an additive morbidity of vascular dementia and AD, the course of vascular dementia has been found to be generally more rapid than AD. Vascular dementia is generally considered to be the most common dementia entity after AD.
1.3 Lewy Body Dementia
Lewy bodies are spherical inclusions located in the cytoplasm of neurons which characteristically contain a protein known as α-synuclein. Originally Lewy bodies were considered to be a prominent feature of Parkinson’s disease and were not associated with late life dementia. However, new staining techniques in the 1990s indicated that Lewy bodies in the brain stem and the cortex commonly occur in dementia patients studied postmortem. Estimates indicate that as many as 15 to 25 percent of dementia patients manifest Lewy bodies at the time of demise (McKeith et al. 1996). However, in the great majority of these cases, Lewy bodies exist together with neuropathologic manifestations of AD and/or vascular dementia. When this mixed neuropathology occurs, the clinical manifestations of the dementia are those of AD and/or vascular dementia, and there is no distinctive Lewy body dementia clinical syndrome.
In approximately 4 percent of all dementia cases coming to autopsy, Lewy bodies occur in the brain in the absence of other dementia disorders. In these cases, a classical Lewy body dementia (LBD) clinical syndrome occurs. This syndrome is marked by three core features, any or all of which may be present: (a) a relatively fluctuating clinical course, with variability in cognition; (b) the presence of vivid, well formed, visual hallucinations; and (c) the presence of Parkin-sonian features. Another important aspect of LBD is sensitivity to neuroleptic (antipsychotic) medications, particularly those known to be associated with Parkinsonian side effects (Ballard et al. 1998). In general, the onset of LBD is more acute than the onset of AD, and the course of LBD appears to be shorter than that of AD.
1.4 Frontotemporal Dementias
These are a diverse group of disorders for which various classification schemata have been developed. For example, biomolecular categorizations have been developed recently which are extremely useful in categorizing one subgroup of frontotemporal demen-tias, those with frontotemporal dementia with Parkinsonism linked to chromosome 17 (FTDP-17) (Foster et al. 1997). The FTDP-17 dementias are characterized by mutations which alter tau, the protein responsible for neurofibrillary changes such as those found in AD. Another frontotemporal dementia type marked by abnormal tau accumulation is Pick’s disease. In Pick’s disease there is an intracellular inclusion known as the Pick body which contains tau accumulations as well as other substances. Other forms of frontotemporal dementia are classified mainly on the basis of their clinical presentation. They include semantic dementia, progressive aphasia, and motor neuron disease dementia.
The core clinical features of the frontotemporal dementias have been described in part as disinhibition, decreased insight, apathy, disorganization, lack of personal hygiene, mental rigidity, and hyperorality (Brun et al. 1994, Kertesz 2000). In addition, aphasia and other language disturbances are frequently present. Like the basic pathologic disturbances, these behavioral manifestations are very heterogenous and occur on a clinical spectrum.
In general, the frontotemporal dementias occur in adults at a younger age than AD. Some believe that these conditions are very common in patients who are institutionalized for mental illness.
1.5 Other Progressive Dementias
Various other diverse conditions produce progressive dementia. In general, these other conditions are believed to be considerably less common than the dementias which have been reviewed more specifically in the preceding sections.
Perhaps the most notable of these other dementias are the prion dementias, of which the most frequent is Creutzfeldt-Jakob disease (CJD). Prion dementias are believed to result from a self-propagating conformational change in a protein, known as the prion protein. This prion, a proteinaceous infective agent, can be acquired either spontaneously or through transmission of biologic materials containing the prion, including brain, pituitary glandular substance, and corneal lens transplants. Spontaneous CJD occurs in approximately one person per million population worldwide. Recently, a form of transmissible CJD arose in Britain as a result of transmission of a prion disease in cattle to humans. This ‘epidemic’ which has affected dozens of persons in Britain and, more recently, several persons on the European continent, has resulted in the destruction of the entire British cattle herd and a change in dietary habits of tens of millions of persons in Europe and elsewhere. Consequently, although the prion dementias remain rare conditions in humans, the threat of the spread of these proteinaceous infections, has caused somewhat justified alarm, in particular in Europe, but also through-out the world.
Other relatively uncommon progressive dementias include progressive supranuclear palsy, and corticobasilar degeneration. These conditions, like some of the frontotemporal dementias, are marked by abnormal tau protein and resultant neurofibrillary accu-mulations.
There are also many progressive dementias associated with broader pathologic spectra, of which dementia is only one element. Among these are the dementia of Down’s syndrome, the dementia of normal pressure hydrocephalus (NPH), and the dementia of Huntington’s disease. Some of these dementias are very closely related to AD dementia. For example, persons with Down’s syndrome universally develop an Alzheimer’s type neuropathologic and clinical picture at a relatively early age in comparison with persons with classical AD. Similarly, brain biopsy studies have recently demonstrated that many elderly persons with NPH who manifest a dementia component to the clinical syndrome, show Alzheimer’s-type pathology, specifically neuritic plaques, upon brain tissue examination (Golomb et al. 2000). As is the case for AD, the percentage of NPH dementia patients with biopsies demonstrating neuritic plaques increases with dementia severity, rising from about 20 percent of patients with MCI to about 75 percent of patients in stage 6 and 7 of the global deterioration scale (see Table 1). These studies indicate that the gait disturbance which is characteristic of NPH appears to be associated with dilated cerebral ventricles (i.e., expansion of the fluid filled cavities in the brain), whereas the dementia component of NPH is probably related to concomitant AD.
2. Dementias With Variable Prognoses
Numerous diverse physiologic disturbances can pro-duce dementias which may be acute or chronic, reversible or permanent. These diverse conditions include infectious conditions, toxins, tumors, brain trauma, endocrine and metabolic disturbances, nutritional disturbances, and medications. For example, infectious conditions which produce encephalitis may cause dementia of viral or bacterial origin. A relatively common example is herpetic encephalitis. Syphilis and acquired immune deficiency syndrome (AIDS), have been associated with dementia. Heavy metal toxicity, such as aluminum toxicity from dialysis, can cause dementia. A space-occupying brain lesion may pro-duce dementia, such as brain metastases resulting from neoplasms. Brain trauma resulting in diffuse brain injury is a source of dementia. Thyroid disturbances, hyponatremia, and other endocrine disorders are known to cause dementia. Vitamin B (cobalamin) deficiency, which is associated with demyelination, is a cause of dementia, as is folate deficiency. Numerous medications can produce dementia or exacerbate a preexisting dementia. Some of the conditions which can produce dementia independently are now believed to be risk factors for AD as well. Examples of the latter include major depression and depression dysphoric disorder, and Vitamin B deficiency.
Bibliography:
- Adams F 1861 The Extant Works of Aretaeus, the Capadocian. Syndenham Society, London
- Ballard C, Grace J, McKeith I, Holmes C 1998 Neuroleptic sensitivity in dementia with Lewy bodies and Alzheimer’s disease. Lancet 351: 1032–3
- Braak H, Braak E 1991 Neuropathological stageing of Alzheimer-related changes. Acta Neuropathologica 82: 239–59
- Braak H, Braak E 1996 Development of Alzheimer-related neurofibrillary changes in the neocortex inversely recapitulates cortical myelogenesis. Acta Neuropathologica 92: 197–201
- Brun A, Englund E, Gustafson L, Passant U, Mann D M A, Neary D, Snowden J S 1994 Clinical and neuropathological criteria for frontotemporal dementia. Journal Neuro Neurosurg Psychiatry 57: 416–8
- Buttini M, Orth M, Bellosta S, Akeefe H, Pitas R E, Wyss-Coray T, Mucke L, Mahley R W 1999 Expression of human apolipoprotein E3 or E4 in the brains of Apoe mice −/− isoform-specific effects on neurodegeneration. Journal of Neuroscience 19: 4867–80
- Esquirol J E D 1838 Des maladies mentales. Balliere, Paris
- Foster N L, Wilhelmsen K C, Sima A A F, Jones M Z, D’Amato C, Gilman S 1997 Frontotemporal dementia and Parkin-sonism linked to chromosome 17: consensus conference. Ann Neurol 41: 706–15
- Galen 1833 De symptomatum differentis liber (Translation). In: Kuhn C G (ed.) Opera Omnia. Knobloch, Leipzig, Germany, Chap. VII, pp. 200–1
- Golomb J, Wisoff J, Miller D C, Boksay I, Kluger A, Weiner H, Salton J, Graves W 2000 Alzheimer’s disease comorbidity in normal pressure hydrocephalus: prevalence and shunt response. J Neurol Neurosurg Psychiatry 68: 778–81
- Hachinski V C, Lassen N A, Marshall J 1974 Multi-infarct dementia: A cause of mental deterioration in the elderly. Lancet 2: 207–10
- Henderson A S, Jorm A F 2000 Definition and epidemiology of dementia: A review. In: Maj M, Sartorius N (eds.) Dementia, WPA Series, Evidence and Experience in Psychiatry. Wiley, Chichester, UK, Vol. 3, pp. 1–33
- Kertesz A 2000 Behavioral and psychological symptoms and frontotemporal dementia (Pick’s disease). International Psychogeriatrics 12(Suppl. 1): 183–7
- McKeith I G, Galasko D, Kosaka K, Perry E K, Dickson D W, Hansen L A, Salmon D P, Lowe J, Mirra S S, Byrne E J, Lennox G, Quinn N P, Edwardson J A, Ince P G, Bergeron C, Burns A, Miller B L, Lovestone S, Collerton D, Jansen E N H, Ballard C, de Vos R A I, Wilcock G K, Jellinger K A, Perry R H 1996 Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB). Neurology 47: 1113–24 National Institute on Aging Alzheimer’s Association Working
- Group 1996 Apolipoprotein E genotyping in Alzheimer’s disease. Lancet 347: 1091–5
- Poirier J, Baccichet A, Dea D, Gauthier S 1993 Cholesterol synthesis and lipoprotein reuptake during synaptic remodel-ling in hippocampus in adult rats. Neuroscience 55: 81–90
- Reisberg B, Ferris S H, de Leon M J, Crook T 1982. The global deterioration scale for assessment of primary degenerative dementia. American Journal of Psychiatry 139: 1136–9
- Reisberg B, Pattschull-Furlan A, Franssen E, Sclan S G, Kluger A, Dingcong L, Ferris S H 1992 Dementia of the Alzheimer type recapitulates ontogeny inversely on specific ordinal and temporal parameters. In: Kostovic I, Knezevic S, Wisniewski H, Spilich G (eds.) Neurode elopment, Aging, and Cognition. Birkhauser, Boston, MA, pp. 345–69
- Reisberg B, Franssen E H, Hasan S M, Monteiro I, Boksay I, Souren L E M, Kenowsky S, Auer S R, Elahi S, Kluger A 1999 Retrogenesis: Clinical, physiologic and pathologic mech-anisms in brain aging, Alzheimer’s and other dementing processes. European Archives of Psychiatry and Clinical Neuroscience 249(Suppl. 3): 28–36
- Reisberg B, Franssen E, Shah M A, Weigel J, Bobinski M, Wisniewski H M 2000 Clinical diagnosis of dementia: A review. In: Maj M, Sartorius N (eds.) Dementia, WPA Series, Evidence and Experience in Psychiatry. Wiley, Chichester, UK, Vol. 3, pp. 69–115
- Rush B 1973 An account of the state of mind and body in old age. In: Rush B (ed.) Medical Inquiries and Obser ations. Dobson, Philadelphia, PA, Vol. 2, pp. 311
- Selkoe D J 1997 Alzheimer’s disease: Genotypes, phenotype and treatments. Science 275: 630–1
- Tomlinson B E, Blessed G, Roth M 1970 Observations on the brains of demented old people. Journal of Neurological Science 11: 205–42
- Xu J, Chen S, Ahmed S H, Chen H, Ku G, Goldberg M P, Hsu C Y 2001 Amyloid-β peptides are cytotoxic to oligodendrocytes. J Neurosci 21, RC118: 1–5
ORDER HIGH QUALITY CUSTOM PAPER

Always on-time

Plagiarism-Free

100% Confidentiality