
View sample cancer research paper on leukemia. Browse other research paper examples for more inspiration. If you need a thorough research paper written according to all the academic standards, you can always turn to our experienced writers for help. This is how your paper can get an A! Feel free to contact our writing service for professional assistance. We offer high-quality assignments for reasonable rates.
History of Leukemia
The term cancer has been in use for over 1500 years; however, it was not until the nineteenth century that the first cases of leukemia were reported. The first breakthrough came in the 1600s with the development of the primitive compound microscope by Anton van Leeuwenhoek, who went on to identify human red blood cells; this was followed by descriptions of white cells and lymphocytes in the late eighteenth century. At the beginning of the nineteenth century, there were reports of patients with irregularities of the blood, but it was almost 100 years after the identification of white blood cells that leukemia was first reported as a distinct clinical entity (Bennett, 1845; Virchow, 1845). Two terms were used to describe this new disease: Virchow introduced leukhemia (white blood) while Bennett preferred leucocythaemia or white cell blood. However, although their research demonstrated that white cells have different types of nuclei, neither could explain how or why these diseases arose. In 1849, Virchow observed that more than one type of the disease existed; these were described as splenic and lymphatic, the former associated with splenomegaly and the latter with large lymph nodes and cells in the blood that resembled those in the lymph nodes (Virchow, 1849). He also proposed his cellular theory of the origin of leukemia, which has been fundamental to present-day understanding of the condition. With respect to childhood leukemia, it appears that the first recorded case was in 1850; interestingly, it was not the acute form that is most commonly seen today, but chronic myeloid leukemia. The acute form of the disease was not described until 1857. Controversy surrounded the claims that leukemia was a separate disease entity but as the number of case reports increased along with pathological and clinical details, it gradually became accepted in its own right as a distinct disease. Concurrently, speculation about the determinants of this cancer also began to increase.
Academic Writing, Editing, Proofreading, And Problem Solving Services
Get 10% OFF with 24START discount code
More breakthroughs occurred in 1868 when bone marrow was discovered to be the source of blood formation and changes in the bone marrow were identified in cases of leukemia. Alongside these discoveries, it was observed that non-nucleated red blood cells formed from nucleated red blood cells in the bone marrow, and that the bone marrow also produced white blood cells. Neumann in 1878 went on to classify leukemia as a disease of the bone marrow and added the myelogenous subtype of leukemia to the splenic and lymphatic types already described. Unfortunately, it took almost 20 years for these theories to be accepted.
Improvements in microscopy and the development of methods to differentiate between cell types allowed the classification of leukemia to be simplified into myeloid (arising from granulocytes in the bone marrow) and lymphoid (arising from lymphocytes, nongranular cells). Ehrlich, who was instrumental in developing this classification, also identified a primitive cell that he described as an ancestral cell in the hematopoietic system, from which other cells in the distinct cell lineages were derived; this may also have been the earliest report of a stem cell (Ehrlich, 1880). In 1900, Naegeli described a new cell type, the myeloblast, which was shown to be an ancestor of the granulocyte. The lymphoblast was also shown to be the cell in the lymphoid line that produced lymphocytes, thus confirming that the cell lineages were distinct. It was from this point onward that the presence of primitive myeloblasts and lymphoblasts in the circulating blood formed a classic diagnosis of acute leukemia. Several years later, monocytic leukemia was described; and by 1913 it was possible to classify leukemia as chronic lymphocytic, chronic myelogenous, acute lymphocytic, myeloblastic, monocytic, or as erythroleukemia.
Significant advances in laboratory techniques over the last 100 years have underpinned progress in the classification of leukemia, which in turn has enabled the development of improved treatments. Thus in 1948, it was shown for the first time that it was possible to obtain temporary remission in patients with acute leukemia following treatment with folate antagonists. In 1962, acute lymphoblastic leukemia was the first systemic malignancy to be cured by chemotherapy. However, despite advances in classifying leukemia subtypes, all acute leukemias in children were grouped together until the late 1960s. It was only the observation that acute myeloid and acute lymphoid leukemias responded differently to treatment that enforced the use of the new technologies to distinguish between them. Today, it is reported that over 70% of cases of acute lymphoblastic leukemia diagnosed in children under 15 years are cured, although improvements have been more limited in adults. However, while much insight has been gained into classification, diagnosis, and treatment of leukemia, unfortunately the same cannot be said about our understanding of the causes.
Leukemia Classification
Leukemias are a heterogeneous group of disorders arising from the unregulated proliferation of a clone of immature hematopoietic cells that have been trapped at an early stage of differentiation and are unable to differentiate into mature functional blood cells. The ensuing clones accumulate in the bone marrow and spill over into the peripheral blood, examination of which enables the cell of origin to be determined. The different subtypes of leukemia are classified according to this presumed cell of origin, along with the clinical behavior, such that patients with acute disease, where the clone of leukemic cells is highly malignant, tend to have a poorer prognosis than those diagnosed with a chronic form, where the clone is of low malignancy. However, clinical outcome is not only dependent on the nature of the leukemic clone, but also on how far the disease has progressed by the time the symptoms are recognized, the diagnosis made, and the treatment implemented. If there is a delay in diagnosis it is more likely that the clone will have progressed to the point where additional mutations will have been accrued, including those that confer drug resistance, making treatment more difficult.
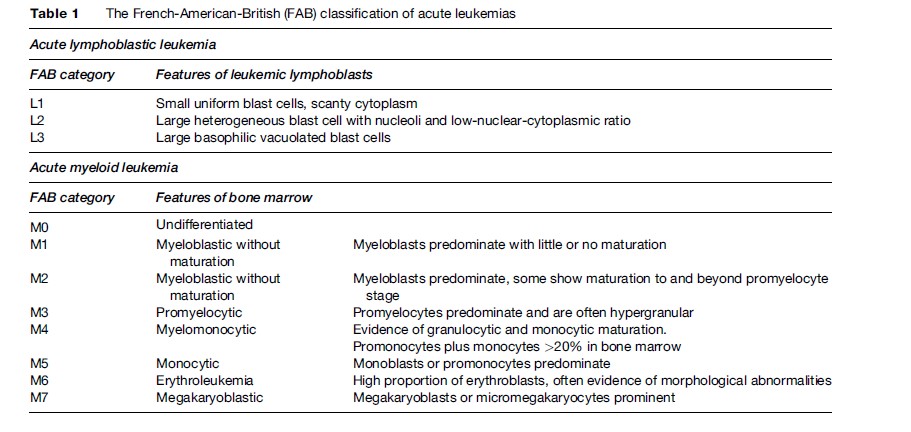
The maturity of the main leukemia cell type seen in the bone marrow and blood is also considered; immature blast-like cells predominate in acute leukemia, whereas more mature cells are seen in chronic leukemia. The acute leukemias are divided into myeloid or lymphoid based on the lineage of the blast cells. Classification of acute leukemia has traditionally been based upon the appearance (morphology) of leukemic cells in a bone marrow aspirate. The French-American-British cooperative group described different variants of acute lymphoblastic leukemia (ALL) and acute myeloid leukemia (AML) based on the morphology and immunophenotypes of the malignant cells, which became known as the FAB subtypes L1–L3 and M0–M7 (Table 1) (Bennett et al., 1976). These two subtypes, along with chronic myeloid (granulocytic) leukemia (CML) and chronic lymphocytic leukemia (CLL) account for the majority of clinical diagnoses of leukemia. Other rarer forms include hairy cell leukemia and prolymphocytic leukemia.
Historically, histological examination has been used for the diagnosis and classification of leukemia subtypes. However, molecular biology techniques have become an integral part of the diagnostic process. These include the use of monoclonal antibodies against various cellular antigens and cytochemical, immunophenotypic, and cytogenetic tests to further characterize the leukemic cells, to determine T and B cell monoclonality, and to detect chromosomal abnormalities. Immunophenotyping, carried out by flow cytometry or immunohistochemistry, is pivotal in distinguishing between AML and ALL and between T cell and B cell subtypes of ALL. Some subtypes have such characteristic immunophenotypic features that diagnoses can often be suggested or excluded based purely on these results. Cytogenetic analysis is doubly beneficial and it provides information about chromosomal abnormalities that not only correlate with particular subtypes of ALL but also with prognosis. Recent advances include the increasing popularity of gene expression microarrays for both diagnostic and prognostic purposes.
Descriptive Epidemiology of Leukemia
Unraveling the causes of leukemia is complex. However, descriptive epidemiological studies of disease incidence, prevalence, mortality, and survival in well-defined populations and subgroups can be of tremendous benefit. The frequency (incidence) of leukemia and of its specific subtypes, typically expressed as the number of new cases per 100 000 population per year, is generally obtained from population-based cancer registries, with the total population at risk obtained from national or local census data.
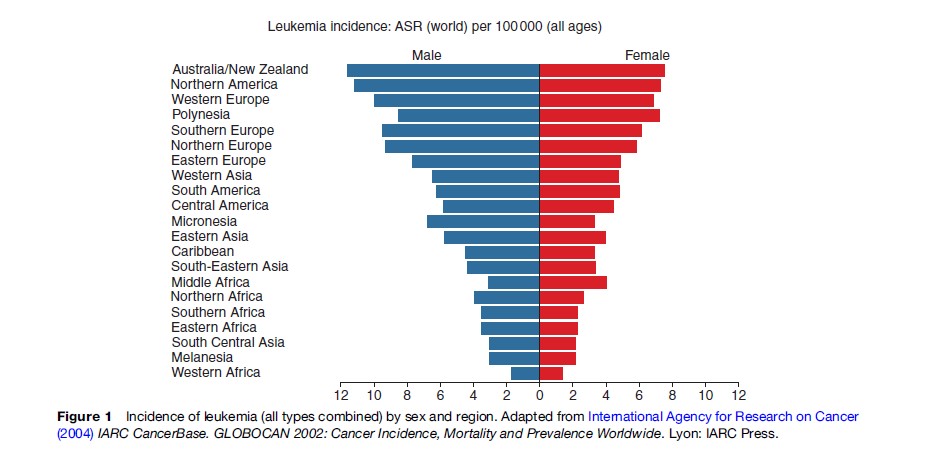
Worldwide, leukemia accounts for around 3% of all cancer diagnoses each year making it the 11th most commonly diagnosed malignancy. As with most other malignant diseases, leukemia is more common in men than women, with more than 56% of all new diagnoses seen in men. Incidence estimates for total leukemia vary around tenfold between countries, ranging from 1.8–12 cases per 100 000 persons per year among men and 1.7–7.8 cases per 100 000 per year among women (Figure 1). The highest rates of leukemia in men are seen in Australia, New Zealand, North America, and Western Europe and the lowest in Western and Middle Africa. For women, leukemia rates are also highest in Australia, New Zealand, and North America, in addition to Polynesia, and are lowest in Western Africa, South-Central Asia, and Melanesia. In the UK, the estimated incidence is high compared with other parts of the world, in particular for men (13.8 per 100 000), in whom it is the ninth most common malignancy. The incidence in women is lower (9.7 per 100 000).
The overall incidence of leukemia and its specific subtypes varies with age. For acute leukemia, the peak incidence in children is between the ages of 2 and 5 years and then a steady decline until the age of 20–24 years. There is a slow increase from ages 30–34 to ages 50–54, followed by a rapid increase with age such that the highest incidence is seen in those over 85 years of age. The peak incidence seen between 2 and 5 years of age is accounted for by ALL, the most common form of leukemia seen in children: 75% of cases of ALL are diagnosed in children under the age of 6 years. In contrast, AML accounts for less than 20% of all leukemias in children, but its incidence increases rapidly with age from around 2 per 100 000 per year in young adults to 10–20 per 100 000 population in those over 65 years of age. Chronic forms of leukemia in children are very rare in all populations and seldom exceed 4% of all leukemia diagnoses; indeed, CLL is rarely seen before the age of 35. However, the incidence of both types of chronic leukemia increases with age.
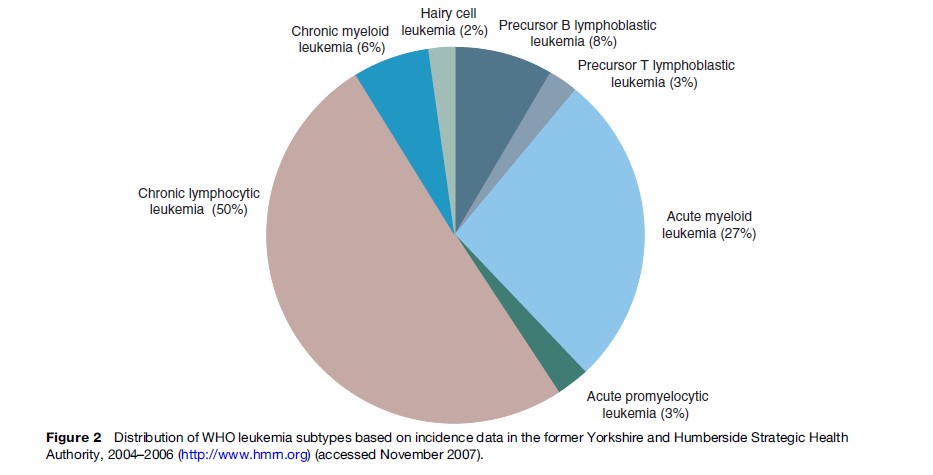
CLL is the most commonly diagnosed form of leukemia, accounting for up to 50% of all diagnoses (Figure 2). It has an annual incidence of two to three cases per 100 000 per year and a male-to-female ratio of 2:1. CML is less frequent (1 per 100 000). There is geographic variation in incidence of the leukemia subtypes; for example while CLL is the most common form of leukemia in the Western world, its incidence in Japan, South America, and Africa is much lower. AML in children is seen more frequently in Asia and among black populations of North America than in Caucasians.
Around 3% of all cancer-related deaths worldwide are attributable to leukemia. In the UK, for example, leukemia is responsible for just under 3% of all cancer-related deaths, and it ranks as the 11th most common cause of death from cancer. Mortality rates for leukemia are slightly higher in men (8.2 per 100 000 per year) than in women (7.2). During the last decade in the UK, there has been a small decrease in the European age-standardized mortality rate for leukemia in women; by contrast, there has been a small increase in the rate for men.
Historically, one of the problems faced when describing patterns of incidence of leukemia and searching for specific risk factors and appropriate treatments has been the heterogeneity of the disease. However, advances in technology that permit more detailed diagnostic analyses but that require less biological material have enabled recent epidemiological studies to investigate the etiology not only of the more common disease subtypes such as AML and CLL but of the rarer subtypes such as hairy cell leukemia (2% of cases) and precursor T-lymphoblastic leukemia (3%) (Figure 2). Since the underlying biological pathways involved in the initiation and progression are likely to be different for each of the distinct disease subtypes, it is important to classify them as individual disease entities when designing new investigative studies. In addition, although the frequency of the rarer subtypes has been, and will continue to be, low, it is important that they are included in future studies and that international consortia are established in order to provide sufficient power to detect any risks.
Etiology Of Leukemia
The etiology of leukemia has not been fully elucidated. However, like most other malignancies, the pathogenesis of leukemia is likely to be a multistep process influenced by a combination of environmental and genetic factors. Carcinogens can accumulate over a longer period of time in adults than in children, so the potential duration of exposure is much shorter in children and it is likely that different exposures are associated with childhood and adult disease subtypes. Exposures acting before birth and early in life have long been thought to be important determinants of the disease in children. Several potential mechanisms by which exogenous agents, including environmental factors and drugs, could impact on childhood leukemia susceptibility have been identified. Areas of interest to date have involved parental smoking before and during the period of conception of the child, parental drug use, exposure to ionizing and nonionizing radiation, folate intake, infant feeding, and most notably the timing and dose of exposure to infectious agents. However, while much progress has been made in the treatment of childhood leukemia, this has not been matched in terms of delineating the underlying causes of the disease (Lightfoot and Roman, 2004).
Several risk factors have been clearly identified for specific leukemia subtypes, however, including ionizing radiation, certain genetic disorders, and exposure to chemotherapeutic drugs, along with other physical and chemical exposures, including benzene. In addition, conditions such as myelodysplastic syndromes, myeloproliferative disorders, pernicious anemia, recovery from aplastic anemia, and paroxysmal nocturnal hemoglobinuria have all been linked with AML. Infection with the human T cell lymphotropic virus type 1 (HTLV1) has been associated with risk of adult T cell leukemia. One other important factor in the pathogenesis of leukemia is the role that chromosomal abnormalities play in the development, classification, and outcome of the disease.
Chromosomal Abnormalities
Most of the original information gathered about chromosomal alterations in cancer has come from studies of leukemia and lymphoma, because it is easier to obtain pure populations of single cell types from peripheral blood or bone marrow samples than from solid tumors such as lung, colon, or breast tumors. Not all leukemias have evidence of chromosomal abnormalities, but several types of chromosomal abnormality are commonly associated with specific leukemia subtypes, such as translocations, deletions and additions, point mutations, and gene amplification.
Acute Myeloid Leukemia
Acute myeloid leukemia (AML) is subclassified according to evidence of lineage differentiation, including AML with recurrent genetic abnormalities, AML with multilineage dysplasia, therapy-related AML and AML not otherwise characterized. The first three all have specific cytogenetic profiles that are important in predicting the leukemic process, especially with respect to response to therapy and survival.
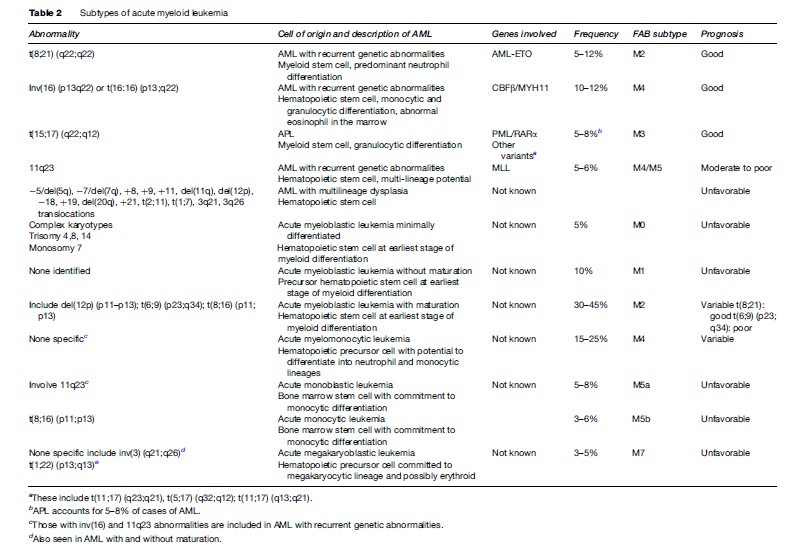
The most common abnormalities seen in AML with recurrent genetic abnormalities include the reciprocal translocations t(8;21) (q22:q22) (AML-ETO), inv(16) (p13q22), t(16;16) (p13:q22) and various translocations involving the 11q23 breakpoint. Many of these tend to occur in younger patients and are generally associated with a good response to therapy, often a high rate of complete remission and a favorable outcome. With respect to 11q23 aberrations, these are frequently seen in two specific clinical subgroups of AML: AML in infants and therapy-related leukemia that can arise after treatment with DNA topoisomerase II inhibitors. Acute promyelocytic leukemia (APL) is a specific form of AML that occurs in adults in mid-life and accounts for 5–8% of all AML cases, with recurrent genetic abnormalities characterized by (15:17) (q22:q12) translocations and abnormal promyelocytes.
In contrast, AML with multilineage dysplasia is more frequently seen in older patients, rarely occurs in children, and is associated with a reduced likelihood of achieving complete remission. It is categorized by an unfavorable cytogenetic profile, often involving additions and deletions of major segments of selected chromosomes including deletions of 5q, 7q, 11q, 12p, and 20q, gain of chromosomes 8, 9, 11, 19, and 21, loss of chromosomes 5, 7, and 18, specific translocations t(2;11), t(1;7), and trans- locations involving 3q21 and 3q26. Deletions involving 5q and/or 7q are associated with a higher incidence of multidrug resistance and poor response to therapy.
Therapy-related AML (t-AML) occurs following cytotoxic chemotherapy and/or radiation therapy and is predominantly associated with balanced translocations involving 11q23, in particular t(9;11), t(11;19), and t(6;11), although others including t(8;21), t(3;21), inv(16), t(8;16), and t(6;9) have also been observed. Less is known about the AML not otherwise classified group, which includes acute myeloblastic leukemia minimally differentiated, acute myeloblastic leukemia with and without maturation, acute myelomonocytic leukemia, acute monoblastic and acute monocytic leukemia, and acute megakaryoblastic leukemia. Some of these, such as acute myeloblastic leukemia without maturation, are quite common, comprising 30–45% of AML cases, and prognosis varies depending on the chromosomal abnormalities identified. More detail about the specific types of AML is given in Table 2.
Acute Lymphoblastic Leukemia
Most cases of ALL (over 80%) arise from malignant primitive precursors of B lymphocytes, while the others are T cell leukemias. ALL is predominantly a disease of children. The immunological subtypes include common, pre-B-ALL, null, and B and T phenotypes. Common ALL is the most frequent, accounting for 50% of all cases of childhood ALL. The segregation by age of these different subtypes may account for the marked prognostic differences between infants, children, and adults (Greaves, 1999). The cytogenetic abnormalities are generally grouped as high hyperdiploidy (over 50 chromosomes per nucleus instead of the normal 46), hyperdiploidy (47–50 chromosomes), hypodiploidy (fewer than 46 chromosomes), pseudodiploidy (46 chromosomes but with structural or numerical changes), and translocations. All of these are used in designing the treatment of ALL in children. Over 200 genes have been found to be involved in chromosomal translocations in ALL, but many are rare; those most commonly associated with childhood ALL are MLL, TEL, and AML1, all of which can fuse with over 15 other genes and, in the case of TEL and AML1, with each other.
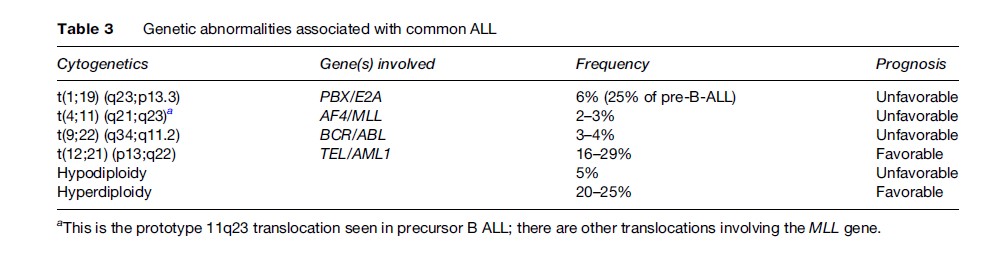
One of the most favorable subtypes of ALL is precursor B-ALL, in which treatment can achieve complete remission in about 95% of children and 60–85% of adults. However, in adult precursor B-ALL, the cases are generally less well characterized genetically; the poor prognostic abnormality t(9;22) (q34;q11.2) (BCR-ABL) is seen in 25% of cases and translocations involving the MLL gene at the 11q23 locus, associated with a poor prognosis regardless of age, are also more common in adults than in children. Conversely, those chromosomal alterations that confer a good prognosis, such as high hyperdiploidy and the t(12;21) translocation, are less frequently seen in adults. Details of the common genetic alterations seen in B-ALL along with the associated prognosis are given in Table 3. Other abnormalities also exist, including del (6q), del(9p), del(12p), hyperdiploidy less than 51, near triploidy, and near tetraploidy, which are all associated with an intermediate prognosis.
Precursor T-ALL, originating from a precursor T lymphoblast, accounts for approximately one in six childhood ALL diagnoses and one in four adult ALL diagnoses. In a third of cases, it is characterized by translocations involving 14q11.2, 7q35, and 7p14–15 loci, in combination with a number of partner genes such as MYC(8q24.1), TAL1(1p32), – RBTN1(11p15), RTBN2 (11p13), HOX11(10q24), and LCK(1p34.3–35). The deletion del(9p) is also seen in around 30% of cases.
Chronic Myeloid Leukemia
Chronic myeloid leukemia (CML) is a clonal myeloproliferative disorder originating from a pluripotent bonemarrow stem cell. It is characterized by a reciprocal translocation between chromosomes 9 and 22, t(9;22) (q34;q11) (BCR-ABL) that results in the Philadelphia chromosome (der(22q)). This translocation occurs in over 95% of all cases, not only in neutrophil precursors but also erythroblasts, megakaryocytes, and some B lymphocytes. It results in the production of a chimeric protein with increased tyrosine kinase activity. The remaining cases of CML either have variant translocations involving a third or even a fourth chromosome, in addition to chromosomes 9 and 22, or cryptic translocations involving 9q34 and 22q11. While t(9;22) translocations have been identified in both CML and adult ALL, albeit only in a minority of cases, those seen in CML arise from different break points from those found in most ALL patients. Both translocations have tyrosine kinase activity but the fusion protein seen in ALL has higher activity, which may account for the fact that ALL is a much more aggressive disease. Development of new treatments for CML has also been focused on the presence of the t(9;22) translocation: Chemical agents have been generated that block the tyrosine kinase activity of the abnormal BCR-ABL protein. Allogeneic bone marrow transplant remains the only curative therapy.
Chronic Lymphoblastic Leukemia
Over 98% of all chronic lymphoblastic leukemia (CLL) diagnoses involve a neoplastic clone of B lymphocytes; although the malignant cells appear morphologically mature, they are actually arrested at an early stage of B cell development. The remaining cases arise from T lymphocytes. Abnormal karyotypes are associated with 80% of cases, with 13q14 deletions observed in up to 50% of cases. Trisomy 21 and deletions at 11q22–23 are also found in 20% of cases; the latter also occurs with somatic mutations in the other allele. Additional abnormalities seen include deletions at 6q21 (5% of cases) and 17p13 (p53 locus) (10% of cases) and expression of p53 in 10% of cases. Many of the alterations seen in CLL appear to contribute to outcome; trisomy 21 correlates with an aggressive clinical course, whereas the 13q14 abnormalities are associated with long survival. Both 11q22–23 deletions and TP53 abnormalities are associated with poor survival. Inherited genetic abnormalities in the ataxia-telangiectasia gene have also been associated with an increased risk of developing CLL.
Genetic Factors
In addition to the chromosomal abnormalities described previously, numerous constitutional genetic defects and disorders have also been associated with risk of leukemia, in particular acute leukemia in children. The most notable association is that between acute leukemia and Down’s syndrome (trisomy 21), in which the incidence of acute lymphoblastic leukemia (ALL) and acute myeloid leukemia (AML) is increased by 20to 30-fold, while acute megakaryoblastic leukemia (AMKL) is 500 times more common in persons with Down’s syndrome than in others (Hasle et al., 2000). The underlying biological mechanisms for these associations are unclear, but progress has been made following the discovery of an exclusive mutation of GATA1, a transcription factor involved in hematopoiesis, in children with both Down’s syndrome and AMKL. However, advances in the molecular understanding of this form of leukemia have not been matched by basic epidemiological information about its incidence. While it appears thus far that ALL is equally common in children with and without Down’s syndrome, the type of AML seen in children with Down’s syndrome is different from that seen in other children. Moreover, the attention concentrated on AMKL has somewhat obscured the fact that ALL, and not AML, remains the most common form of leukemia in children with Down’s syndrome. A UK-based study has been designed to address the paucity of epidemiological information and to further our understanding of the specific characteristics that may contribute to acute leukemia susceptibility (www.cdss.org.uk).
As well as trisomy 21, several other rare inherited genetic disorders have also been associated with predisposition to leukemia; these include Bloom, Klinefelters, and Li-Fraumeni syndromes, Fanconi anemia, ataxia-telangiectasia, and neurofibromatosis. Fanconi anemia (FA) is a rare condition seen in around 1 in 200 000 live births, and although it is usually inherited as an autosomal recessive trait, it is genetically heterogeneous, and includes an Xlinked form. While AML is the most common leukemia associated with FA, reported in approximately 10% of cases, cases of T cell and B cell ALL have also been observed. The association of both germ-line and somatic mutations in the FA pathway with AML susceptibility has been considered. Although current data provide little evidence of a role for inherited FA gene mutations in AML susceptibility, reduced levels and in some cases a total absence of FA proteins has been observed in both AML cell lines and primary AML samples. AML that arises in people with FA is associated with a high incidence of myelodysplasia and other unfavorable clinical features, and it is possible that this is due to abnormal susceptibility to DNA damage, global genomic instability, or a combination of both. Ataxia-telangiectasia (AT), another autosomal recessive disorder, has been associated with a number of leukemia subtypes, including childhood T cell ALL. Deletion of the gene associated with AT, ataxia-telangiectasia mutated (ATM), has been reported in CLL. Neurofibromatosis type 1 and Noonan syndrome are also both related to leukemia in infants and young children, and they are particularly linked to juvenile myelomonocytic leukemia and other types of myeloproliferative disorder.
Environmental Exposures
The list of suspected chemical, physical, and biological agents associated with increased leukemia risk continues to increase, although the evidence for some is often limited or even contradictory. However, several exposures are widely accepted as known risk factors for leukemia.
A good model by which to study chemical leukemogenesis is provided by therapy-related AML (t-AML). Therapy-related leukemias can be categorized into two main groups, depending on the source of exposure. One group is associated with exposure to chemotherapeutic alkylating agents and is characterized by chromosomal abnormalities involving chromosomes 5 and 7. The other is linked to exposure to topoisomerase II inhibitors and is typified by balanced translocations involving the MLL gene at 11q23. Since infant leukemia is also characterized by 11q23 translocations, it is possible therefore that in utero exposure to topoisomerase inhibitors could also be important in infant leukemia risk. However, the list of topoisomerase inhibitors is extensive and includes specific chemotherapeutic agents such as etoposide but also benzene metabolites (derived from cigarette smoke and pollution for instance), bioflavonoids, herbal medicines, anthraquinone laxatives, podophyllin resin, quinolone antibiotics, pesticides, and most phenolic chemicals and their metabolites. Furthermore, topoisomerase II inhibitors have been found in specific fruits, tea, coffee, wine, soy, and cocoa and it is quite likely that many other chemicals that have the ability to inhibit this enzyme have yet to be identified. The ever-increasing list of chemicals that can act as topoisomerase II inhibitors means that investigating the association between this group of compounds and leukemia risk is complex, since many of them will have effects on other biological pathways. Therefore, trying to ascertain which, if any, is the important pathway for disease pathogenesis may not be straightforward.
Ionizing Radiation
Ionizing radiation is a well-known risk factor for leukemia, and AML is one of the most common malignancies associated with exposure. A dose-dependent increase in AML was observed in survivors of the atomic bomb explosions in Japan, with a peak incidence 5–7 years after exposure to neutrons and gamma rays. There have also been reports of increased incidence of leukemia in workers exposed to the highest levels of radiation following the accident at the Chernobyl nuclear processing plant in 1986. Both AML and ALL have been identified in other casualties of the Chernobyl disaster who were exposed to beta-particles at a young age. AML is commonly seen in people who have received high-dose spinal radiation for ankylosing spondylitis. A particular area of concern is the increased potential for radiation-induced second malignancies such as AML, since patients with cancers such as prostate and breast cancer tend to be treated with radiation therapy at younger ages and survive for longer. The most pertinent issue for AML is that the time interval between exposure and development of the disease is typically only a few years. There are difficulties in applying the results of early studies to assess the risk of leukemia following radiotherapy because the radiotherapy protocols have evolved considerably. Complex mechanistic models have been developed to enable prediction of radiation-induced cancer risks, and these should help by optimizing treatment plans to minimize the incidence of leukemia following radiotherapy.
It is generally accepted that the fetus and young child may be more susceptible to the effects of ionizing radiation than the adult, with modern concern revolving mainly around the importance of dose and gestational age at the time of exposure. Epidemiological evidence that prenatal exposures were involved in the development of childhood leukemia was first provided by the Oxford Survey of Childhood Cancers over 50 years ago, when an association between diagnostic radiography of mothers during pregnancy was related to the subsequent development of leukemia and other cancers in their children (Stewart et al., 1956). Good data also exist for paternal occupational exposure to ionizing radiation; in 1990, Gardner and colleagues reported that the recorded external dose of whole-body ionizing radiation to fathers during their time at Sellafield was associated with the development of leukemia in their offspring. Their results suggested that the highest risks were seen in those with highest accumulated ionizing radiation doses prior to conception, either over their total duration of exposure or during the preceding 6 months. Case–control studies carried out in the vicinity of other nuclear establishments where workers were, on average, exposed to much lower doses were unable to confirm these findings and the results of other large-scale studies are contradictory. The paucity of historical human data on this topic, coupled with the fact that workers nowadays are routinely exposed to much lower doses, means that additional human data on this topic are unlikely to accrue.
Benzene
Benzene is a widespread environmental contaminant, still found in glues, solvents, and gasoline. It is a product of incomplete hydrocarbon combustion and is present in traffic exhaust fumes as well as in tobacco smoke. The greatest risk for exposure to high doses of benzene occurs in the workplace, although the most common exposure to lower doses of benzene occurs in the general environment. Exposure to benzene can either be through inhalation or skin absorption with inhalation of contaminated air the primary route of exposure.
It is well established that exposure to benzene is associated with an increased risk of AML, although the latency period following benzene exposure is not as accurately defined as that for radiation or chemotherapy induced AML. Initially, increased risks of leukemia, predominantly AML, were reported among workers with high levels of benzene exposure in the chemical, shoemaking, and oil-refining industries, whereas more recently studies have focused on workers with lower exposure. While the underlying mechanisms relating benzene exposure to AML are unclear, it is thought to involve damage to specific regions of DNA, which subsequently results in chromosome abnormalities, including rearrangements and deletions. Chromosomal changes commonly seen in AML, including monosomy 7, trisomy 8, and t(8;21), have been identified in blood cells of workers in Shanghai who had been heavily exposed to benzene, and DNA methylation patterns similar to those seen in AML have also been observed following exposure to low-level airborne benzene. Furthermore, 5q deletions are also common in AML patients who have been exposed to benzene or alkylating agents. In addition, it has been proposed that benzene metabolites may act as DNA topoisomerase II inhibitors, and induce abnormalities at 11q23.
Evidence from both epidemiological and laboratory animal studies supporting the association between benzene exposure and carcinogenesis has led to measures to limit occupational and environmental exposure. In the United States, the Environmental Protection Agency (EPA) has enforced limits on concentrations of benzene in drinking water, with the ultimate aim being to abolish it altogether. The National Institute of Occupational Safety and Health (NIOSH) and the Occupational Safety and Health Administration have limited occupational exposures to benzene to 1 part per million during an average working day; they also recommend personal protective equipment such as respirators. Importantly, changes in policy that restrict smoking in public places will also reduce environmental exposure to benzene.
Summary
Leukemia is a complex and heterogeneous group of disorders. Understanding the determinants of leukemia has been aided by advances in immunological techniques, which have helped to identify chromosomal abnormalities that have significance not only for classification of the different subtypes of leukemia, but also for prognosis and treatment. A number of acquired genetic abnormalities, including trisomy 21, are associated with a higher risk of specific leukemia subtypes and much may be learned from focusing on those groups. While specific exposures relating to AML risk have been identified, such as benzene, ionizing radiation, and chemotherapeutic alkylating agents, less is known about risk factors for other leukemia subtypes including ALL. The largest area of interest for ALL, particularly with respect to childhood ALL, remains the potential etiological role of the timing and dose of exposure to infectious agents. The success story to date has been the treatment of childhood leukemia, because over 80% of all cases now survive for long periods and may be considered cured.
Bibliography:
- Bennett JH (1845) Case of hypertrophy of the spleen and liver in which death took place from suppuration of the blood. Edinburgh Medical and Surgical Journal 413–423.
- Bennett JM, Catovsky D, Daniel MT, et al. (1976) Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. British Journal of Haematology 33: 451–458.
- Ehrlich P (1880) Methodologische Beitrage zur Physiologie und Pathologie der verschisdenen Formen der Leukocyten. Zeitschrift Klinische Medizinische 1: 553–558.
- Greaves M (1999) Molecular genetics, natural history and the demise of childhood leukaemia. European Journal of Cancer 35: 173–185.
- Hasle H, Clemmensen IH, and Mikkelsen M (2000) Risks of leukaemia and solid tumours in individuals with Down’s syndrome. Lancet 355: 165–169.
- International Agency for Research on Cancer (2004) IARC CancerBase. GLOBOCAN 2002: Cancer Incidence, Mortality and Prevalence Worldwide. Lyon, France: IARC Press.
- Lightfoot TJ and Roman E (2004) Causes of childhood leukaemia and lymphoma. Toxicology and Applied Pharmacology 199: 104–117.
- Stewart A, Webb J, Giles D, and Hewitt D (1956) Malignant disease in childhood and diagnostic irradiation in utero. The Lancet 2: 447.
- Virchow R (1845) Weisses Blut. Froriep’s Notizen 36: 151–156.
- Virchow R (1849) Zur pathologischen Physiologie des Blutes. IV. Farblose, pigmentierte und geschwantze nicht spezifische Zellen im
- Archives of Pathology Anatomy and Physiology 2: 587–598. Epidemiology and Genetics Unit, University of York (n.d) Yorkshire and Humberside Strategic Health Authority, 2004–2006.
- Greaves M (2006) Infection, immune responses and the aetiology of childhood leukaemia. Nature Reviews Cancer 6: 193–203.
- International Agency for Research on Cancer (2001) Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: World Health Organization.
- Piller G (2001) Leukaemia – A brief historical review from ancient times to 1950. British Journal of Haematology 112: 282–292.
- Pui CH (2006) Childhood Leukemias. 2nd edn. Cambridge, UK: Cambridge University Press.
- https://www.hmrn.org/ – Haemotological Malignancy Research Network.
- https://ukccs.org/ – United Kingdom Childhood Cancer Study.
More Cancer Research Paper Examples:
ORDER HIGH QUALITY CUSTOM PAPER

Always on-time

Plagiarism-Free

100% Confidentiality