
View sample cancer research paper on cancer and senescence. Browse other research paper examples for more inspiration. If you need a thorough research paper written according to all the academic standards, you can always turn to our experienced writers for help. This is how your paper can get an A! Feel free to contact our writing service for professional assistance. We offer high-quality assignments for reasonable rates.
Increased Risk Of Cancer Development With Aging
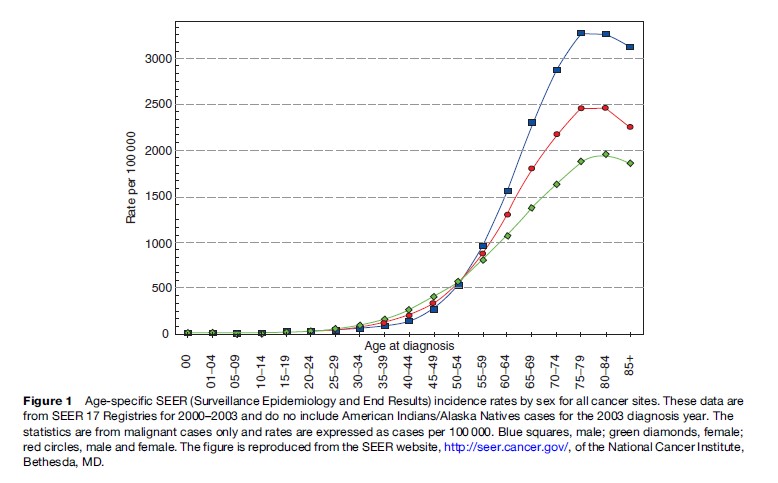
The adult human body consists of approximately 60 trillion cells. The rate of proliferation and growth of this vast number of cells are under strict regulation and are highly coordinated. However, in rare cases, a few cells in a particular organ escape this tight regulation of growth, start uncoordinated proliferation, acquire invasive and metastatic properties, and become cancerous. The incidence of cancer rises exponentially with age in humans (Ries et al., 2006). As shown in Figure 1, until age 35, the incidence of cancer is 0.2% in the general population. This incidence rises to about 1.5% at ages 50–60. The maximum incidence of cancer, which is close to 5%, occurs between ages 70 and 80. Therefore, cancer tends to develop after 50 years of age, which is halfway from the 100th year, the maximum lifespan of humans. In addition, women tend to have twice the incidence of cancer at age 30–50 than men, although this trend reverses after age 50. After age 85, there tends to be a slight decline in the incidence of cancer.
Academic Writing, Editing, Proofreading, And Problem Solving Services
Get 10% OFF with 24START discount code
The genetic basis of this increased incidence of cancer with age is unclear. Recent progress in aging and cancer research suggests that errors accumulated in DNA during each round of replication, together with several other genetic and epigenetic factors, are involved in the increased risk of cancer with advanced age. In addition to genetic factors such as the accumulation of mutations in DNA, epigenetic factors, such as methylation of DNA and acetylation or deacetylation of histones, which do not affect DNA sequence per se, but change the expression of genes, also contribute to various diseases, including cancer (reviewed in Jones and Laird, 1999). Other factors that may contribute to the age-dependent increase in cancer incidence are DNA repair defects (reviewed in Lombard et al., 2005), impaired mitochondrial function (reviewed in Taylor and Turnbull, 2005), and the decrease in immune surveillance (reviewed in Burns and Leventhal, 2000). Here we focus on the role of an altered cellular phenotype, known as cellular senescence, which is associated with aging in culture and in vivo.
Cellular Senescence And Accumulation Of Senescent Cells In Aging Tissues
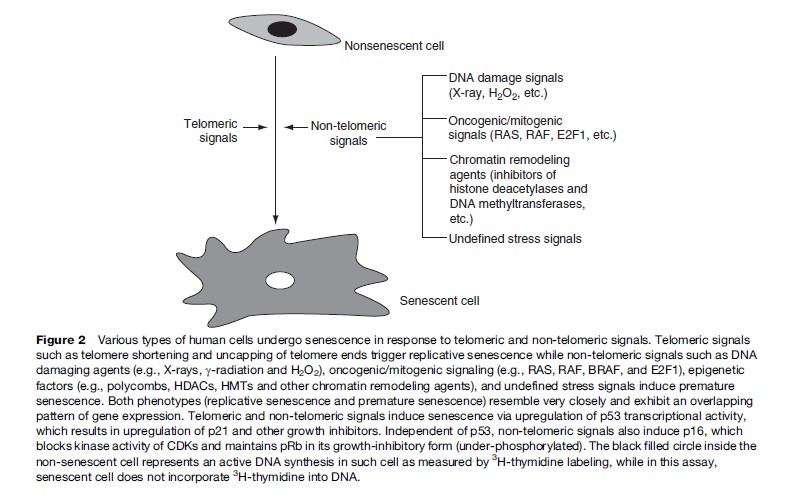
Normal human cells irreversibly arrest growth with a large and flat cell morphology after a limited number of cell divisions. This process is termed cellular senescence and was first described by Hayflick and colleagues in cultured human fibroblasts (Hayflick and Moorhead, 1961). Since eukaryotic cells have linear chromosomes, each chromosome shortens from the ends, or telomeres, during every round of cell division due to the biochemistry of DNA replication. It is thought that senescence in culture may also reflect an aging process in vivo (reviewed in Itahana et al., 2004). It is generally accepted that human fibroblasts and other types of cells senesce because they eventually acquire one or more short dysfunctional telomeres. In addition to telomeric signals, recent evidence suggests that cells also undergo senescence in response to acute signals that are potentially oncogenic. These signals include inappropriate activation or expression of oncogenes, strong mitogenic signals, direct DNA damage, and chromatin remodeling agents (Figure 2) (reviewed in Campisi, 2005; Dimri, 2005). Senescence induction by these nontelomeric signals may be much more important in age-related diseases such as cancer.
Using senescence-associated b-gal (SA-b-gal) as a bio- marker to detect senescent cells (Dimri et al., 1995), it has been shown that at least in some tissues, senescent cells accumulate in the human body with advanced age (Itahana et al., 2004). For example, senescent cells accumulate in aging human skin, primate retina, and liver. Senescent cells have also been detected at sites of age-related pathology, such as benign hyperplastic prostate and atherosclerotic lesions. If senescent cells do accumulate in vivo, the natural question is what is the cell’s role in vivo? In the late 1990s, based on theory of evolutionary antagonistic pleiotropy, Campisi proposed a hypothesis of senescence being a double-edged sword (Campisi, 1996). The theory of evolutionary antagonistic pleiotropy states that genes and processes that are beneficial to an organism early in life are detrimental later in life (Kirkwood and Austad, 2000). When this theory is applied to senescence, Campisi proposed that senescence, by virtue of its tumor suppressor nature, must be beneficial to us early on in life but actually may be bad in later life by contributing to an agedependent increase in cancer incidence (Campisi, 1996, 2005). As discussed below, there is accumulating evidence to support this hypothesis.
Possible Contribution Of Senescence To Oncogenesis
Elegant studies from Campisi’s laboratory have shown that, at least in some cases, the accumulation of senescent cells provides a pro-oncogenic signal (Campisi, 2005).
It has been found that compared to nonsenescent cells, senescent cells secrete a high level of matrix metalloproteinases, growth factors, and inflammatory cytokines (Campisi, 2005). The inappropriate expression of these factors can influence oncogenesis in multiple ways, most notably by altering the stromal environment, making it such that cancer cells can thrive and prosper in it. Indeed, it was shown that senescent fibroblasts stimulate proliferation of premalignant epithelial cells in culture and facilitate tumorigenic conversion of preneoplastic epithelial cells in a mouse model (Krtolica et al., 2001). Recently, it was shown that senescent fibroblasts upregulate vascular endothelial growth factor (VEGF) expression and secretion (Coppe et al., 2006). The secreted VEGF by senescent fibroblasts was shown to increase invasiveness of human umbilical vascular endothelial cells in culture and cause increased vascularization of tumors in vivo (Coppe et al., 2006). Recently, senescent prostate fibroblasts were also shown to enhance proliferation of neoplastic prostate epithelial cells through paracrine mechanisms (Bavik et al., 2006).
The detailed characterization of other secretory molecules responsible for pro-oncogenic activities of senescent fibroblasts remains to be investigated. It is also important to note that most of these studies employed fibroblasts only; at present, not much is known about the pro-oncogenic contribution of other senescent cell types. Moreover, at present, direct evidence showing the accumulation of senescent cells in the vicinity of a malignant tumor is lacking. Nonetheless, it seems reasonable to assume that the accumulation of senescent cells in aged tissues could facilitate cancer development by creating a matrix environment that is more conduit to cancer progression.
Senescence Acts As An Initial Barrier To Cancer Development
On the bright side, senescence has been convincingly shown to be a tumor suppressor mechanism and an initial barrier to cancer development. It is very well established that cellular senescence is regulated by tumor suppressor pathways (Dimri, 2005). In particular, functional pRb (retinoblastoma tumor suppressor protein) and p53 pathways are required for the genesis and maintenance of the senescent phenotype (Dimri, 2005). Inactivation of these tumor suppressors results in the bypass of senescence and propensity to cell immortalization. Due to essentially irreversible growth arrest characteristics and the requirement for p53 and pRb functions, cellular senescence is considered a potent tumor suppressor mechanism.
The importance of cellular senescence in cancer is underscored by the fact that the first step in creating an in vitro model of human cancer involves the abrogation of cellular senescence (reviewed in Boehm and Hahn, 2005). These studies show that a combination of SV40 large T, small t, hTERT (human telomerase catalytic subunit), and H-RAS is able to transform a variety of normal human cell types such as fibroblasts, embryonic kidney cells, mammary epithelial cells, ovarian epithelial cells, and endothelial cells (Boehm and Hahn, 2005).
Recent published reports from various laboratories clearly show that cellular senescence is an important anticancer defense and acts as an initial barrier to cancer development in vivo. It was found that the expression of BRAF oncoprotein, activated H-RAS, or inactivation of the tumor suppressor PTEN induces an acute senescence response in vivo, and, as a result, tumor progression is blocked at a very early stage (Braig et al., 2005; Chen et al., 2005; Collado et al., 2005; Michaloglou et al., 2005). Tumor progression was facilitated only by additional mutations that caused bypass of senescence. Accordingly, malignant tumors that eventually arose in these systems were largely devoid of biomarkers of senescence including SA-b-gal. In the Michaloglou et al. study, it was found that the benign nevi in human patients, which express the oncogenic form of BRAF, by and large are made of senescent melanocytes, as these cells express markers of senescence such as SA-b-gal and p16 tumor suppressor. Patients with malignant melanomas showed no presence of senescent cells. In the transgenic model of lung cancer progression by K-Ras, it was shown that premalignant tumors but not malignant tumors contained senescent cells (Collado et al., 2005). In another study, oncogene induced senescence (OIS) was shown as an initial barrier in lymphoma development in the Eu-N-Ras transgenic mouse model (Braig et al., 2005). Finally, Chen et al. showed that PTEN inactivation per se leads to the induction of premature senescence in vitro and in vivo, which is p53-dependent. It was also shown that early-stage benign human prostate cancer samples but not late-stage prostate cancer samples contained senescent cells (Chen et al., 2005). Collectively, these studies suggest that senescence induction, in particular OIS, acts as an initial barrier to cancer progression in vivo. Although it is yet to be shown, it is very likely that senescence induction in response to other potential oncogenic signals such as DNA damage, mitogenic signals and chromatin remodeling signals, and telomere attrition also act as an antioncogenic response.
Senescence In Cancer Treatment
A number of in vitro studies have demonstrated that chemotherapeutic drugs act in part by inducing premature senescence (Roninson, 2003). In addition, recent studies provide compelling evidence that senescence can also be induced in vivo by chemotherapeutic drugs (Schmitt et al., 2002; te Poele et al., 2002; Roberson et al., 2005). In a mouse model, Schmitt et al. showed that a chemotherapy drug CTX (cyclophosphamide) is able to engage a senescence program when apoptosis is inhibited by Bcl2 overexpression. In another study, te Poele et al. stained newly sectioned archival breast tumors from patients who had undergone chemotherapy regimens for senescence markers. It was found that the normal tissues adjacent to the tumors were devoid of SA-b-gal and p16 protein, while 15 out of 36 (41%) tumors stained positive for the SA-b-gal marker and had high expression of p16 (te Poele et al., 2002). Similarly, induction of premature senescence by chemotherapy treatment of human lung cancer was also recently demonstrated (Roberson et al., 2005).
Thus, chemotherapeutic drugs can induce a senescence-like stage in vivo, implying that these drugs may work via induction of a senescence-like stage. Another important regulator of senescence is telomerase and its target telomeres. More than 90% of tumors contain readily detectable telomerase activity; in principle, telomerase can be targeted to cause telomere shortening and induce apoptosis or senescence in precancerous and tumor cells (Shay and Wright, 2002). Indeed, several telomerase inhibitors are known to induce senescence and/or apoptosis in tumor cells (Dimri, 2005). Thus, senescence induction by chemotherapeutics clearly contributes to the antioncogenic activity of these compounds.
Concluding Remarks
The relationship between aging and cancer is well recognized; however, the underlying mechanisms are not fully understood. As discussed here, senescence is a common thread that connects cancer and aging. Senescence can act both ways: it can protect an organism from cancer or it can facilitate cancer development, depending on when and where it occurs. Although the latter role of senescence may be more significant during aging, it is also possible that the failure of cells to undergo senescence due to defects in senescence checkpoints contributes to late life cancers. It is conceivable that, due to genetic and epigenetic factors, in some cells, senescence checkpoints are compromised, and that these cells, when challenged with potential oncogenic signals, fail to invoke a senescence response and undergo oncogenesis. On the other hand, as discussed earlier, the presence of senescent cells could be detrimental, when present in the vicinity of cells that are halfway from being fully malignant. It is important to note that although senescent cells do accumulate with age, the fraction of senescent cells is always minor when compared to the vast overall number of nonsenescent cells in various organs, even in the aged tissues. Nobody can predict how many senescent cells are needed to facilitate cancer development. On the other hand, it is predictable that if senescence checkpoints are compromised, there is a good chance that the cells will become cancerous. In other words, senescence may be guilty of aiding and abetting cancer unknowingly in some cases, but by and large, it protects us from developing cancer. In summary, a better understanding of the facts is just what the jury needs to convict or acquit this fascinating phenotype, when it comes to one of the most frightening diseases of our time, cancer.
Bibliography:
- Bavik C, Coleman I, Dean JP, Knudsen B, Plymate S, and Nelson PS (2006) The gene expression program of prostate fibroblast senescence modulates neoplastic epithelial cell proliferation through paracrine mechanisms. Cancer Research 66: 794–802.
- Boehm JS and Hahn WC (2005) Understanding transformation: Progress and gaps. Current Opinion in Genetics and Development 15: 13–17.
- Braig M, Lee S, Loddenkemper C, et al. (2005) Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 436: 660–665.
- Burns EA and Leventhal EA (2000) Aging, immunity, and cancer. Cancer Control 7: 513–522.
- Campisi J (1996) Replicative senescence: An old lives’ tale? Cell 84: 497–500.
- Campisi J (2005) Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell 120: 513–522.
- Chen Z, Trotman LC, Shaffer D, et al. (2005) Crucial role of p53dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 436: 725–730.
- Collado M, Gil J, Efeyan A, et al. (2005) Tumour biology: Senescence in premalignant tumours. Nature 436: 642.
- Coppe JP, Kauser K, Campisi J, and Beausejour CM (2006) Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. Journal of Biological Chemistry 281: 29568–29574.
- Dimri GP (2005) What has senescence got to do with cancer? Cancer Cell 7: 505–512.
- Dimri GP, Lee X, Basile G, et al. (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proceedings of the National Academy of Science USA 92: 9363–9367.
- Hayflick L and Moorhead PS (1961) The serial cultivation of human diploid cell strains. Experimental Cell Research 25: 585–621.
- Itahana K, Campisi J, and Dimri GP (2004) Mechanisms of cellular senescence in human and mouse cells. Biogerontology 5: 1–10.
- Jones PA and Laird PW (1999) Cancer epigenetics comes of age. Nature Genetics 21: 163–167.
- Kirkwood TB and Austad SN (2000) Why do we age? Nature 408: 233–238.
- Krtolica A, Parrinello S, Lockett S, Desprez PY, and Campisi J (2001) Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proceedings of the National Academy of Science USA 98: 12072–12077.
- Lombard DB, Chua KF, Mostoslavsky R, et al. (2005) DNA repair, genome stability, and aging. Cell 120: 497–512.
- Michaloglou C, Vredeveld LC, Soengas MS, et al. (2005) BRAFE600associated senescence-like cell cycle arrest of human naevi. Nature 436: 720–724.
- Ries LAG, Harkins D, Krapcho M, et al. (eds.) SEER Cancer Statistics Review, 1975–2003. Bethesda, MD: National Cancer Institute.
- Roberson RS, Kussick SJ, Vallieres E, Chen SY, and Wu DY (2005) Escape from therapy-induced accelerated cellular senescence in p53-null lung cancer cells and in human lung cancers. Cancer Research 65: 2795–2803.
- Roninson IB (2003) Tumor cell senescence in cancer treatment. Cancer Research 63: 2705–2715.
- Schmitt CA, Fridman JS, Yang M, et al. (2002) A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell 109: 335–346.
- Shay JW and Wright WE (2002) Telomerase: A target for cancer therapeutics. Cancer Cell 2: 257–265.
- Taylor RW and Turnbull DM (2005) Mitochondrial DNA mutations in human disease. Nauret Review Genetics 6: 389–402.
- te Poele RH, Okorokov AL, Jardine L, Cummings J, and Joel SP (2002) DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Research 62: 1876–1883.
- Campisi J (2005) Senescent cells, tumor suppression, and organismal aging: Good citizens, bad neighbors. Cell 120(4): 513–522.
- Dimri GP (2005) What has senescence got to do with cancer? Cancer Cell 7(6): 505–512.
- Fossel MB (2004) Cells, Aging, and Human Disease. New York: Oxford University Press.
- Ries LAG, Harkins D, Krapcho M, et al. (eds.) SEER Cancer Statistics Review, 1975–2003. Bethesda, MD: National Cancer Institute.
- Weinberg RA (1998) One Renegade Cell – How Cancer Begins. London: Weidenfeld and Nicolson.
- http://seer.cancer.gov – National Cancer Institute, SEER.
- http://www.nature.com/nature/focus/senescence/index.html – Nature web focus: Senescence: Cells, ageing and cancer.
More Cancer Research Paper Examples:
ORDER HIGH QUALITY CUSTOM PAPER

Always on-time

Plagiarism-Free

100% Confidentiality