
Sample Genetic Studies Of Behavior Research Paper. Browse other research paper examples and check the list of research paper topics for more inspiration. If you need a research paper written according to all the academic standards, you can always turn to our experienced writers for help. This is how your paper can get an A! Feel free to contact our custom research paper writing service for professional assistance. We offer high-quality assignments for reasonable rates.
The phrase ‘methodology in genetic studies of behavior’ is intended to cover any scientific analysis of behavioral phenomena that includes biometric genetic information. Human beings have multiple thousands of genes—specific DNA sequences on specific areas of the chromosomes—and each gene represents a specific sequence of amino acids that creates RNA for the formation or coding of specific proteins. Different versions of a gene appear as a pair of alleles at a specific position or locus, and a specific collection of alleles are labeled a genotype. While over 99 percent of all genes in humans are identical to the corresponding genes in all other humans, the other 1 percent are important because they are thought to lead to individual differences in both genotypes. Human beings exhibit multiple thousands of observed behaviors and any well-defined subset of these behaviors is labeled a phenotype. In these terms, the methods of genetic analysis of behaviors are used to clarify and improve our understanding of the relationships among particular variations or combinations of genotypes and particular variations or combinations of phenotypes.
Academic Writing, Editing, Proofreading, And Problem Solving Services
Get 10% OFF with 24START discount code
Genetic methods and analyses have roots in the nineteenth century, specifically in Charles Darwin’s well-known ideas on evolution and Gregor Mendel’s work on the mechanisms of heredity in plants. Many techniques used in genetic analyses of behavior were initiated in the agricultural sciences to better control the size and health of plants and animals, further developed in the medical sciences to understand and control sources of disease, and are now routinely used in large-scale family studies to better understand many behavioral phenomena. Most of the latter methods described here stem from the classical nineteenth century work of Sir Francis Galton on the measurement of individual differences in physical and mental characteristics (Johnson et al. 1985).
In this research paper, we discuss some historical background, we focus on contemporary methodological issues, and we emphasize the methodological unity of most current genetic approaches. We do not deal with important contemporary issues about the use of genetic information (i.e., genetic testing and screening, ethical use of genetic information, human cloning, genetically altered food, etc.).
1. Classical Methods In Genetics And Behavioral Studies
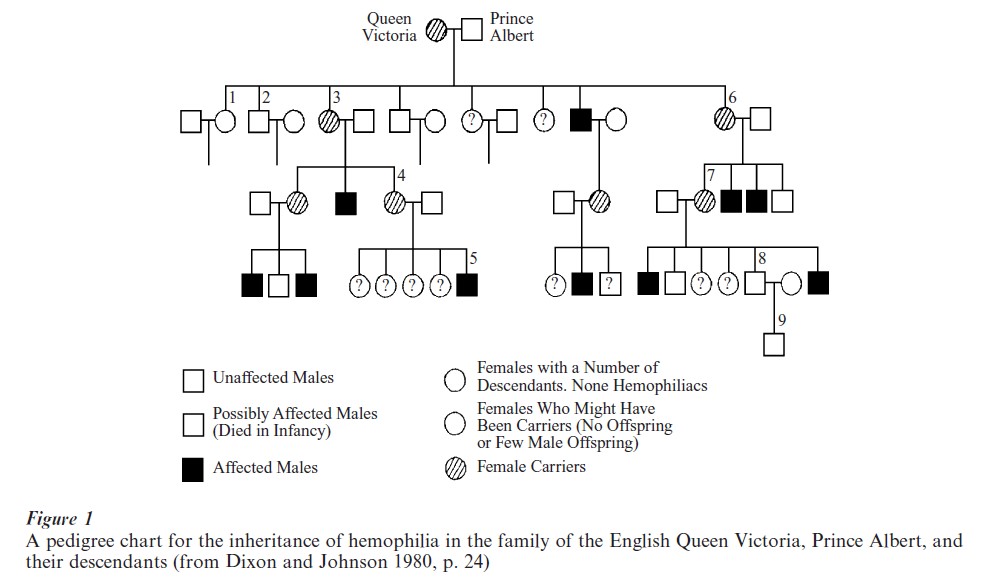
From a historical point of view, human genotypes have been very difficult to measure and have often been treated as unobserved or latent attributes. Analyses of family trees or ‘pedigrees’ were among the first scientific evidence provided to clearly establish that aspects of human behavior are influenced by genetic variation. Figure 1 is a chart of a family tree or ‘pedigree’ from Galton of the inheritance of hemophilia in the family of the English Queen Victoria, Prince Albert, and their descendants. The pedigree method has been used to establish whether given disorders (e.g., hemophilia) run in families and, if so, what kind of inheritance (dominant or recessive, autosomal or sex-linked) best fits the observed pattern, for example hemophilia was found to be a sex-linked characteristic carried on the X chromosome. In another well-known example, clinical research on mental retardation, especially research showing the effects of inbreeding and the genetic cause of Down syndrome, has established the role for genetic regulation of impaired intellectual functioning (for references see Dixon and Johnson 1980, McArdle and Prescott 1996).
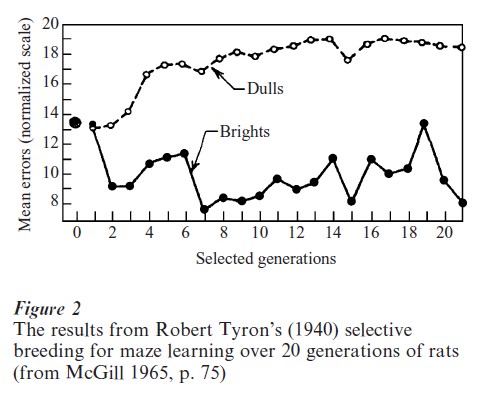
Findings from clinical disorders led researchers to investigate whether genetic factors might regulate aspects of behavior within the normal range of variation. Early evidence for genetic effects on normal behaviors came from selective-breeding studies with non-human animals. Figure 2 is a plot of results from the well-known selective-breeding studies of Tolman, Jeffress, and Tryon (Tryon 1940 as reported by McGill 1965). In these studies a heterogeneous stock of rats was tested on learning tasks in an automatic maze. The observed performance in these learning tasks led the experimenters to classify each rat into one of two groups—‘dull’ or ‘bright’—within which the rats were allowed to mate and produce offspring for the next generation. After accounting for some deleterious effects (e.g., inbreeding and regression), these investigators showed that after successive selection of relatively few generations this behavioral selection led to distinct strains of ‘maze-bright’ and ‘maze-dull’ rats. With no change in rearing environments, these experiments provided direct evidence for the role of genetic components in learning.
Galton also initiated large-scale studies based on family resemblance. He studied physical features (e.g., height) in parents and children and reported the law of ‘filial regression (reversion) towards mediocrity’ better known as ‘regression to the population mean.’ He also studied intellectual achievement and reported that individuals born to higher social classes attained higher educational and social standing than individuals born to lower classes. These family studies were not controlled experiments, so scientific inferences about the separate impacts of changing genotypes and changing environments remained obscure. Selective breeding experiments were impossible with human populations for ethical reasons. Nevertheless, experimental concepts derived from animal research gradually led to creative use of a novel class of methods based on correlations observed among atypical groups of families—‘twin studies,’ ‘adoption,’ ‘half-sibs,’ and other ‘genetically informative’ family groupings were discussed in the early twentieth century by many researchers including Thorndike, Siemans, Merriman, and Fisher (see Spector et al. 2000).
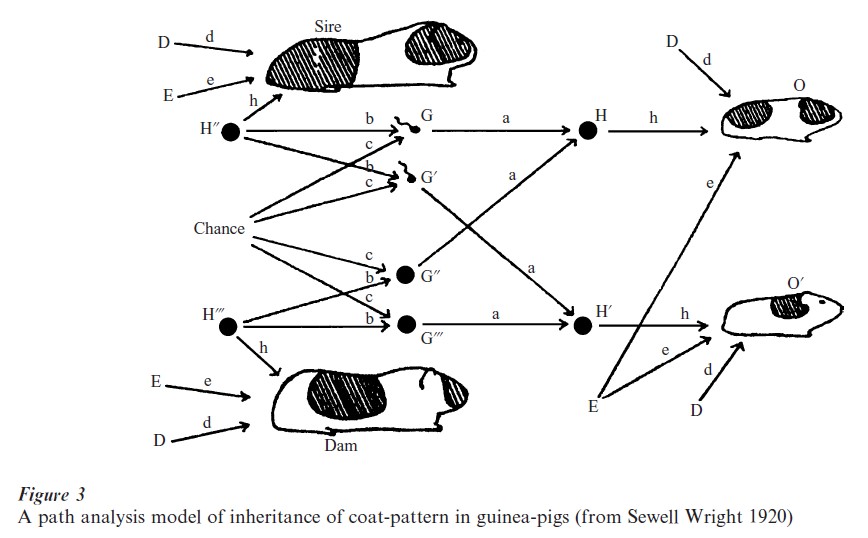
Multivariate statistical methods were developed to accommodate unobserved or latent genotypes. Figure 3 is a path diagram from the classical work of geneticist Wright (1920) showing a complex system of influences. Here Wright suggested the environmental factors (E ) were mixed with the heredity influences (H ) together with ‘Chance’ factors (and those labeled D), and all were required to account for the ‘coatpattern’ of a guinea-pig litter. More complex models of inheritance were defined by the quantitative rules of Mendelian inheritance and the factors of the correlation decomposition of the statistician R. A. Fisher and later by many other including R. B. Cattell, and J. Jinks and D. Fulker (see Falconer and Mackay 1996).
Advanced versions of these genetic methods are now widely used in behavioral research. The search for specific genetic abnormalities linked to behavioral disorders continues at a rapid pace in research from the Human Genome Project (e.g., Strachan and Read 1995). Genotypes are now directly related to behaviors using experimental techniques based on direct manipulation of the genome in model organisms, as well as statistical techniques of linkage and association (Elston 1998). In advanced statistical studies, substantial progress has been made in the use of mathematical and statistical techniques for studies of family resemblance based on latent genotypes and latent phenotypes (McArdle and Goldsmith 1990, Neale and Cardon 1992). In the next four sections we highlight contemporary issues in these methodologies and present some issues for future research.
2. Contemporary Experimental Methods With Measured Genes
The last decade of the twentieth century has seen a revolution in molecular genetic methodology. Techniques based on recombinant DNA methods permit direct measurement and experimental manipulation of variations in genotype or gene expression. It is useful to divide these techniques on the basis of whether they primarily use information about a gene’s position within the genome, a gene’s function, a gene’s DNA sequence, or a gene’s protein product to develop our understanding about the genetic influences on behavioral characteristics.
Studies based on positional information are usually aimed at the long-term goal of positional cloning of the gene or genes that influence some trait (Burmeister 1999, Schork and Chakravarti 1996). In positional cloning, one capitalizes on the fact that Mendel’s law of independent assortment does not hold for genes that are physically near each other on the same chromosome, so genes do not assort randomly. Rather they tend to co-segregate within families to a degree that is monotonically and directly related to their physical proximity to each other on a chromosome. A genetic marker is a specific physical location on a strand of DNA. If co-segregation is observed between a genetic marker and a phenotype, it is reasonable to conclude that the marker is physically close or linked to a gene influencing the phenotype. When this cosegregation is examined as an association between markers and phenotypes within families one is generally testing for linkage. When this co-segregation is examined as an association between markers and phenotypes across nominally unrelated individuals one is often testing for linkage disequilibrium (Elston 1998).
There are many types of studies that use positional information to find genes influencing traits in non- human animals (e.g., Lynch and Walsh 1998). Selective breeding studies with measured genes are the conceptually simplest method used to locate genes influencing traits. These studies are based on measurements of the frequencies of alleles at genetic loci in an outbred population. Any loci exhibiting statistically significant changes in allele frequency over generations are either locus influencing the trait or linked to loci influencing the trait.
A more complex but efficient method uses recombinant inbred strains. Once several inbred strains are produced, one animal from each line can be genotyped at a set of marker loci. Because all animals within a strain have the same genotype, it is only necessary to genotype one animal within each strain to know all animals’ genotypes. At each locus of interest, one simply tests whether the different lines (i.e., AA vs. BB genotypes) have different phenotypic outcomes. If significant differences are found, this is evidence that the locus under study either causes variation in the phenotype or is linked to a locus that causes variation in the phenotype. In certain situations, the use of inbred strains can be quite efficient in terms of both genotyping and phenotyping costs. More complex methods involve intercrosses between inbred strains, multipoint analyses with simultaneous uses of multiple markers, multivariate analysis of multiple phenotypes, selective sampling, and other enhancements.
One limitation of all approaches based on positional information is that they only work with existing variation within a gene, that is, they do not tell us what genes are fundamentally important to a phenotype when all individuals within a population have the same alleles at that gene. In one kind of experimental approach, molecular geneticists are now able to alter or remove particular genes and observe the behavioral effects. One of the most common types of induced mutation studies is the development of a ‘knockout’ animal where a specific targeted gene is disrupted or ‘knocked-out.’ Another type of study is based on the use of random insertion clones—expressed sequences tag (EST) acting as a molecular homing device—to disrupt genes throughout the genome. Another type of direct genetic manipulation is referred to as the generation of a ‘transgenic’ model where additional genetic material or a new gene is inserted into an animal. A final approach to studying gene function uses microarrays, small silicon chips, which have been set up to simultaneously measure the amount of expression of literally thousands of genes. This approach is permit study the circumstances under which certain genes come in to play and, in future research, may permit analyses of multiple interactions of measured genes with measured environments.
3. Contemporary Statistical Methods Based On Measured Genotypes In Humans
In human studies, one classical way to find genes that influence behavioral traits is to look for cytogenetic anomalies among rare individuals with respect to those traits (see Dixon and Johnson 1980). If a cytogenetic anomaly affects a large segment of the chromosome (i.e., missing, transposed, or present in extra copies), and it can be detected in the individuals, then it is plausible that the genomic region of the anomaly harbors a gene influencing the trait. While this method may provide a good starting point for further study, most behaviors are not marked by discontinuity, are not related to abnormality, and cannot be examined in this simple way.
In the method known as linkage analysis, associations (e.g., correlations) between transmission of a measured allele and some measured phenotype are studied within families (Elston 1998). The variety of methods for testing for linkage in human studies is now quite large, but the prototypical study is the sibpair study. Two classes of tests are commonly applied to quantitative genetic information. Neither relies on the assumption of a particular genetic model (Elston 1998) and both are based on allele sharing at marker loci (Lander and Schork 1994). Covariance-based tests are based on standard statistical principles and are commonly used (Allison et al. 2001). Under the null hypothesis of no linkage between a marker and a quantitative trait locus (QTL), there will be no association between the number of alleles a sib-pair shares identical by descent (IBD) and their degree of phenotypic similarity for the traits under study. In contrast, when there is linkage between the marker and the QTL, there will be a positive association between (a) the number of alleles a pair shares IBD and (b) the degree of phenotypic similarity of the pair. Thus, by evaluating whether there is evidence of positive covariance between IBD and phenotypic similarity one can test for linkage. Another class of tests is based on inferring such association by observing a departure of the ‘marginal’ distribution of IBD from its null expectation in samples of sib-pairs that have been selected on the basis of their intra-pair phenotypic similarity or dissimilarity.
One of the key issues in both human and nonhuman genetics, particularly linkage analysis with large numbers of candidate genes, is the determination of an appropriate threshold for statistical significance given the enormous amount of multiple testing conducted (Lander and Schork 1994). Although no definitive consensus now exists, probability or p-values greater than about 10− are rarely taken as compelling evidence for a linkage effect. Reports of p-values smaller than 10− are common and are often persuasive. However, as in any statistical model, these pvalues are calculated under various untested assumptions (e.g., distributional assumptions, accuracy of genotyping, accuracy of phenotyping, independence of observations) and the test procedures may lead to empirical Type I error rates which exceed (or fall below) nominal alpha levels (e.g., Allison et al. 1999). It follows that small p-values alone should not be taken as convincing evidence for a linkage finding in the absence of additional evidence. Researchers looks for careful documentation of the statistical robustness of the finding (e.g., bootstrap and sensitivity analyses), replication in an independent data set, or confirmation through independent methods such as gene knockouts in animal models.
Perhaps the chief limitation of linkage analysis in the context of human studies is low statistical power. For example, to detect a QTL explaining 5 percent of the variance in a phenotype would usually require that one study, or at least screen, thousands of sibling pairs. The reasons for this low power are manifold, but chief among them is the fact that in linkage analysis in humans, one is generally not examining the direct effect (in terms of mean displacement) of different alleles at a QTL, but rather the variance explained by the QTL (i.e., a variance component). In addition, this is only an indirect estimate based on the observation of IBD scores at a marker allele, and these alleles are only partially correlated with IBD scores at the QTL. This being the case, initial analyses focus on a simple test of mean differences in phenotype by genotype at a putative QTL; such a test is referred to as an ‘association test’ in the human genetics literature. Under most circumstances, association tests will be enormously more powerful than linkage tests when one has the polymorphisms of the actual QTL in hand. The typically greater power of association tests has led some to suggest that linkage analyses be replaced by association tests, but this position has led to controversy (e.g., Risch and Merikangas 1996).
One special concern comes from the need to control for background correlations or other confounding, usually referred to as admixture or population stratification in the context of human genetic studies. These confounds arise from the fact that the effects of alleles at any one locus may be stochastically dependent upon the effects of alleles present at another locus even if these two loci are not physically linked on the same chromosome. To avoid the inappropriate conclusions that could stem from such confounding, Spielman et al. (1993) developed what is termed the transmission disequilibrium test (TDT). In the TDT, one counts the number of times a particular allele is transmitted to an offspring affected with the disease from a parent that is heterozygous at the marker locus. If a particular allele is transmitted from a heterozygous parent (i.e., a parent that has two different alleles at the candidate locus) more than 50 percent of the time (i.e., the chance expectation) this is evidence that, not only is that allele associated with the disease, but that it is also linked to a locus which causes the disease. This is because, under ordinary circumstances, each allele a parent has at a genetic locus has an equal (50 percent) chance of being transmitted to an offspring, so any difference from this expectation is evidence of both linkage and association. Since then, numerous extensions of the TDT approach have been developed for both qualitative and quantitative traits (e.g., Allison 1997, Allison et al. 1999).
4. Contemporary Statistical Methods Based On Latent Genotypes In Humans
The vast numbers of human genotypes are very difficult to measure and are necessarily treated as latent attributes. This is especially true when a complex of influences and a complex of outcomes are considered. In behavior genetics and genetic epidemiology all statistical methods utilize classical biometric genetic theory to define and specify the relations among the underlying components thought to represent a continuously distributed trait (e.g., Falconer and Mackay 1996). Most models start by asserting that individual genes combine to produce the underlying genotype. Furthermore, these genes are assumed to be: (a) numerous or ‘polygenic,’ (b) additive in effect, (c) segregating independently (i.e., the probability of receiving one gene is independent of the probability of receiving others), (d) not sex-linked (i.e., located on the X or Y chromosomes), and (e) not influencing characteristics used for spousal selection.
These biometric assumptions are important because they lead to statistical expectations about the observed correlations among pairs of relatives. If all resemblance between relatives in a family is due to genetic sources, the correlations between their observed phenotypic scores are expected to be proportional to the correlations between their genotypic backgrounds, and unrelated individuals would be uncorrelated. More specifically, the genotypes of first-degree relatives correlate (on average in a large sample) at 1 2. First-degree relatives include fraternal (also called dizygotic or DZ) twins, ordinary siblings, and parentchild relationships. The genotypes of second-degree relatives (grandparents-grandchildren, aunt-niece, half-siblings) are expected to correlate 1 4, and the third-degree relatives (cousins) at 1 8, etc. Unrelated individuals living together, such as stepsiblings or parents and their adopted children would be expected to have zero genetic correlation. In the special case of identical (monozygotic or MZ) twins, the genotypes are considered identical and correlate 1.0. Of course, the adequacy of inferences from the sampling of people that are related or unrelated is a major concern of behavior genetic researchers.
Nongenetic influences are often considered as well. In some models the similarity in behavior is expected to be correlated with the similarity in relevant environments (e.g., home of rearing or social background). If true, the highest correlations would be found in scores from twins and similar-aged siblings, lower correlations from parents and children, and still lower correlations from related individuals who have not lived together. Thus, under a purely environmental hypothesis, adoptees should be as similar to their adopting parents and siblings as any other children, and related individuals living in different environments would have scores with no correlation. A model that includes both genetic and environmental sources of familial resemblance would produce estimates intermediate between these other two. All such models can include measurement error and the influence of experiences unique to particular individuals, so even MZ twins living together are not expected to have scores that correlate perfectly.
Different family data permit different assessments of the theoretical components. Figure 4(a) illustrates this kind of latent path model for a ‘family’ design. In this model, additive genetic and shared environmental sources of resemblance are confounded, that is, any interpretation of family resemblance cannot be partitioned into separate additive genetic scores (A) and common family environment scores (C ). In the adoption design of Fig. 4(b) these A and C components are (theoretically) separable, and the only source of parent-child resemblance is through cultural transmission, resulting in an estimate of common family effects. By comparing differences in resemblance among family members in the adoptive versus biological family designs, an estimate of the additive genetic (A) contribution to the phenotypic deviation is obtained.
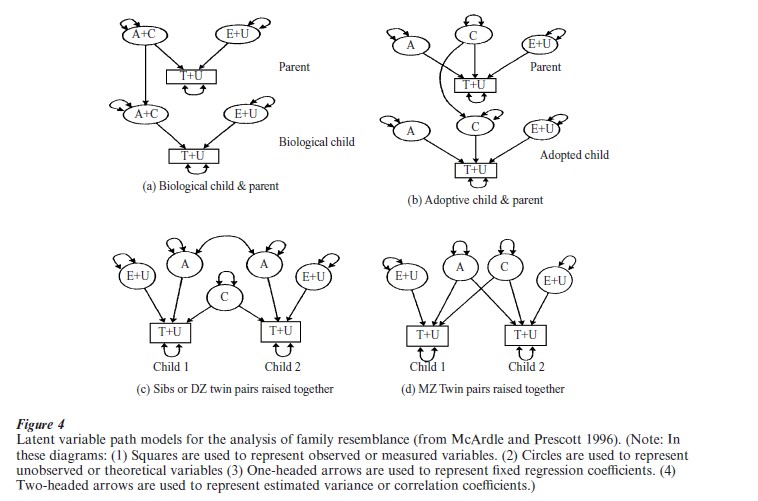
Figures 4(c) and 4(d) show models for resemblance between siblings or fraternal (DZ) twins and identical (MZ) twins. In this kind of model, pairs of DZ twins and pairs of full siblings are assumed to have the same genotypic correlation, even though the siblings can differ widely in chronological age. Unlike the family and adoption models, here we cannot estimate the effects of assortative mating based on the correlation between A and C. In most twin models these correlations are assumed to be zero although fixed estimates from other sources can be inserted. Using these assumptions, the twin design can provide estimates based on the difference between the resemblance between MZ and DZ pairs. The special case of MZ twins raised apart (MZA) is important because, in theory, these data permit a complete separation of these latent components.
Structural equation models developed in several areas of science are used in behavior genetics to organize, estimate, and test aspects of these theories of underlying influences (McArdle and Goldsmith 1990, Neale and Cardon 1992). In some recent models an observed phenotype (P) is often cast as a simple combination of true scores (T ) and error of measurements (W ). Behavioral genetics models go further and include unobserved variables representing the biometric hypotheses: Additive genetic scores (A), nonadditive genetic scores (D), common family environments (C ), other nongenetic (E ) components, and possibly other complex sources. The goal of the structural analysis is independently estimate the total impact (contribution to the variance) of the separate components using the available data from relatives. The statistical basis of the models relies on features common to many scientific analyses—maximum likelihood estimation and likelihood ratio comparisons (e.g., chi-square tests). A sequence of nested statistical tests is routinely used to examine the necessity of each latent component and confidence boundaries can be created.
After specific model coefficients are estimated at a specific value, it is possible to standardize the unobserved variables, assume the latent scores (A, C, and E ) are uncorrelated (orthogonal), and calculate the unobserved genetic variance. In order to compare results for phenotypes with different measurement scales, it has become common to calculate the ‘heritability’ ratio—the unobserved additive genetic varivance divided by the observed phenotypic variance (h2=Va /Vp). This kind of calculation may or may not take into account the common family environment, other genetic factors (dominance, epstasis), and is limited by the sample phenotypic varianvce. In most published research this sample statistic (h2) is reported and the utility of this routine practice has been seriously questioned (e.g., Gottlieb 1997, Wachs 1999, Hooper in Spector et al. 2000).
There has been a great deal of recent progress towards addressing more complex problems and creating methods that permit the study of more complex patterns of influences. Contemporary behavioral genetic methods do include the possibility of evaluating these more complex patterns of influence, including: parent to child environmental transmission of shared effects, further decomposition of the nonshared environments, the correlation between spouses, and the separation of measurement error via multiple phenotypes. The possibility of a correlation between A and C variables is an important feature of contemporary models. These correlations can be meaningful but they often cannot be estimated without more extensive data collections and more complex models. In this recent work, parameters for all effects are estimated by assuming the model parameters are invariant over all the multiple groups of Fig. 4. Other important model features include nonadditive genetic effects such as dominance (nonadditive effects of alleles at the same locus) and epistasis (nonadditive effects of genes at different loci) and the general utility of mean intercepts, subject sampling corrections, and multivariate phenotypes (see McArdle and Goldsmith 1990, Neale and Cardon 1992, McArdle and Prescott 1996).
5. Future Issues In Genetic Methods For Behavioral Studies
As with any methodology, there are both strengths and weakness of the genetic methods discussed here. Direct gene measurement permits usually strong experimental designs—in gene association analyses, the appropriate dependent variable is phenotype and the appropriate independent variable is genotype. However, in most studies the genotype is a nearby marker and not the gene of interest. This produces several unresolved questions that generate skepticism about the value of association tests: (a) Can candidate loci and polymorphisms be identified with sufficient confidence to be useful? (b) Can enough separate polymorphic loci be studied to do genome-wide association studies when candidate loci cannot be confidently identified on an a priori basis? (c) Can confounding by population admixture/stratification be controlled? (d) Will applied investigators conduct association studies with adequate rigor even when the tools are available to them?
The challenge of creating hypotheses about candidate genes must be acknowledged, but this is more a limitation than a flaw of the method. The availability of sufficient markers when one wants to go beyond prespecified candidate genes, the ability to economically assess a large number, and the ability to determine how large that number needs to be are currently being addressed by many researchers. The occasional lack of rigor in the conduct of association studies should not be trivialized, but is a problem that potentially affects all areas of research.
There is ongoing research in trying to understand genes and their role in producing phenotypic effects by studying the more complete gene sequence and geneprotein structure. This latter field is often referred to as proteomics and is one of the most active fields in molecular biology at the beginning of the twenty-first century. These methods will become increasingly important in the future and, moreover, will be combined with studies involving information about gene position, gene function, and behavioral measurements to provide a much richer picture about the way genes work to produce their effects. In these more detailed analyses of measured genes we may also be able to study about how gene-protein functions are stimulated by specific environments.
In a similar vein, the statistical methods for latent genotypes rest on assumptions that may not be reasonable representations of the underlying mechanisms, and some of these assumptions are not testable. Statistical biases arise in adoption studies when the effects of selective placement lead to a correlation between the genotypes of the biological and adoptive parents. In twin data, positive assortative mating leads to underestimates of additive genetic effects and overestimates of downward and common family effects. In addition, the standard twin model assumes that both kinds of twins experience similar environments to an equal degree, but the similarity of MZ pairs is in part due to sharing a more similar common environment, the additive genetic effects will be raised spuriously.
But other modeling assumptions are more fundamental and problematic. For example, the classical models above routinely assume no interaction of the genetic and non-genetic components, initially because these additional components are hard to estimate without detailed measurements of both the genotypes and the environments. Similarly, the concept of genotypes as immutable characteristics with asymmetric direct effects on phenotypes is often challenged. Researchers often suggest that both genes and the environments play an important role in creating constraints on developmental processes (e.g., Waddington 1962, Gottlieb 1997, Wachs 1999).
More complex genetic methodologies are needed to answer these complex questions about complex human characteristics. These are not questions that can be simply answered in terms such as, ‘we have found the gene(s) Q for behavior(s) P,’ or, ‘recent twin research shows P is largely genetic.’ Further answers for the questions raised here will, no doubt, combine the best aspects of both measured and latent genotype approaches. Recent research demonstrates the natural convergence of QTL analysis and latent variable structural models (e.g., Neale in Spector et al. 2000). Newer models are likely to include basic information about the phenotypic outcomes in terms of multivariate measurement models combined with longitudinal dynamic modeling.
A clearer understanding of the methods, models and results from these newer analyses may illuminate the complexity of genetic theory and behavioral theory, as well as the appropriateness of scientific inferences that can be made from such studies. We are given pause when we consider that, even after the entire human genome is accurately mapped, we will still be confronted with an enormously complex set of analytic problems—of linking genotypes to proteins, and proteins to physiology, and physiology to basic levels of behavior, and basic behaviors to more complex ranges, constraints, and the resulting limitations on multiple behaviors. The prior methodological advances have carried us very far, but we now look forward to the future development of novel methods for these monumental tasks.
Bibliography:
- Allison D B 1997 Transmission disequilibrium tests for quantitative traits. American Journal of Human Genetics 60: 676–90
- Allison D B, Heo M, Kaplan N, Martin E R 1999 Development of sibling-based tests of linkage in the presence of association for quantitative traits that do not require parental information. American Journal of Human Genetics 64: 1754–64
- Burmeister M 1999 Basic concepts in the study of diseases with complex genetics. Biological Psychiatry 45: 522–32
- Dixon L K, Johnson R C 1980 The Roots of Individuality: A Survey of Human Behavior Genetics. Brooks Cole, Monterey, CA
- Elston R C 1998 Linkage and association. Genetic Epidemiology 15(6): 565–76
- Falconer D S, Mackay T F C 1996 Introduction to Quantitative Genetics, 4th edn. Longman, London
- Gambaro G, Anglani F, D’Angelo A 2000 Association studies of genetic polymorphisms and complex disease. Lancet 355 (9200): 308–11
- Gottlieb G 1998 Normally occurring environmental and behavioral influences on gene activity: From central dogma to probabilistic epigenesis. Psychological Review 105: 792–802
- Johnson R C, McClearn G E, Yuen S, Nagoshi C T, Ahern F, Cole R R 1985 Galton’s data a century later. American Psychologist 40(8): 875–92
- Lander E S, Schork N J 1994 Genetic dissection of complex traits. Science 265(5181): 2037–48
- Lynch M, Walsh B 1998 Genetics and Analysis of Quantitative Traits. Sinauer Association, Sunderland, MA
- McArdle J J, Goldsmith H H 1990 Some alternative structural equation models for multivariate biometric analyses. Behavior Genetics 20(5): 569–608
- McArdle J J, Prescott C A 1996 Contemporary models for the biometric genetic analysis of intellectual abilities. In: Flanagan D P, Genshaft J L, Harrison P L (eds.) Contemporary Intellectual Assessment: Theories, Tests and Issues. Guilford Press, New York, pp. 403–36
- McGill T E 1965 Readings in Animal Behavior. Holt Rinehart & Winston, New York
- Neale M C, Cardon L 1992 Methodology for Genetic Studies of Twins and Families. Kluwer, Dordrecht, The Netherlands
- Risch N, Merikangas K 1996 The future of genetic studies of complex human diseases. Science 273(5281): 1516–17
- Schork N, Chakravarti A 1996 A nonmathematical overview of modern gene mapping techniques applied to human diseases. In: Mockrin S C (ed.) Molecular Genetics and Gene Therapy of Cardiovascular Disease. Marcel Dekker, New York, pp. 79–109
- Spielman R S, McGinnis R E, Ewens W J 1993 Transmission test for linkage disequilibrium: The insulin gene region and insulin-dependent diabetes mellitus (IDDM). American Journal of Human Genetics 52(3): 506–16
- Spector T D, Sneider H, MacGregor A J (eds.) 2000 Advances in Twin and Sib-pair Analysis. Oxford University Press, London
- Strachan T, Read A P 1995 Human Molecular Genetics. Wiley- Liss, New York
- Wachs T D 1999 Necessary But Not Sufficient: The Respective Roles of Single and Multiple Influences of Individual Development. American Psychological Association, Washington, DC
- Waddington C H 1962 New Patterns in Genetics and Development. Columbia University Press, New York
- Wright S 1920 The relative importance of heredity and environment in determining the piebald pattern of guinea pigs. Proceedings of the National Academy of Sciences 6: 320–32
ORDER HIGH QUALITY CUSTOM PAPER

Always on-time

Plagiarism-Free

100% Confidentiality