
Sample Psychiatric Endocrinology Research Paper. Browse other research paper examples and check the list of research paper topics for more inspiration. If you need a research paper written according to all the academic standards, you can always turn to our experienced writers for help. This is how your paper can get an A! Also, chech our custom research proposal writing service for professional assistance. We offer high-quality assignments for reasonable rates.
The term ‘psychiatrie endocrinienne’ (‘psychiatric endocrinology’) was first coined by Laignel-Lavastine in 1908 at a psychiatric congress in Dijon. Since then hormones always have been viewed as magic bullets for the treatment of a variety of psychiatric conditions and even Sigmund Freud predicted that hormone treatment might one day replace psychoanalysis. The first systematic research in this field was conducted by the Swiss psychiatrist Manfred Bleuler, who attempted to establish a connection between psychopathology and the secretory activity of endocrine glands. Influenced by his mentor, the neurosurgeon Harvey Cushing from Boston, he first directed his interest toward the effect of pituitary-adrenocortical hormones that are excessively secreted in patients with an autonomous adenoma at the pituitary gland. This adenoma produces corticotropin in high amounts unrestrained by the high plasma cortisol concentration that would normally suppress corticotropin release through negative feedback action. Bleuler defined the ‘endocrine psychosyndrome’ and his descriptions of the behavioral changes associated with endocrine dysfunction are still valid today.
Academic Writing, Editing, Proofreading, And Problem Solving Services
Get 10% OFF with 24START discount code
This research paper illustrates how Bleuler’s concept has been translated into hypothesis-driven research and how that has ultimately led to a novel pharmacology that holds the promise of better antidepressant treatments. In addition, some examples are given showing how peripherally monitored changes in hormonal secretion can be viewed as the readout of disturbed central neurobiology. Finally, the current status of several selected hormonal treatments is examined.
1. Stress Hormones
The majority of patients with Cushing’s syndrome, whether this is due to high plasma corticosteroid levels caused by ectopic secretion or high-dosed corticosteroid treatments, develop mood disorders, some with psychotic features. Only in very rare instances do manic or schizophrenic syndromes prevail. Apart from these disturbances in mood and behavior, cognitive impairments are also observed. Cortisol was suspected to account for neuronal loss and damage because the hippocampus, a brain area critical for cognitive function and richly endowed with receptors to which cortisol binds (corticosteroid receptors), was found to be reduced in size in patients with elevated stress hormone secretion. Based on some simplistic concepts, the central actions of cortisol have been described as detrimental to neuronal viability. This impairment, however, is only true when extremely high corticosteroid levels are experimentally induced (Almeida et al. 2000). Under such conditions, which probably mimic the clinical situation only very rarely, glucose uptake by neurons could be reduced, thereby making neurons vulnerable to other insults such as hypoxia or increased neuronal firing rates (e.g., during seizures). Such neuronal damage does not occur in situations of stress-elicited hypercortisolism. On the contrary, corticosteroids are essential to coordinate the organism’s ability to cope with stress and promote the interpretation and storage of novel information, as well as facilitating the extinction of behavior that is not relevant under the immediate highly demanding situation. Significantly, under certain circumstances corticosteroids can act as neuroprotectants rather than as neurotoxicants.
Much of the current confusion related to the excitement about the phrase ‘stress is bad for your brain’ stems from the different functions that the two corticosteroid receptor types, the glucocorticoid (GR) and the mineralocorticoid receptor (MR) may exert. As studies by Ron de Kloet and colleagues (1999) in Leiden, the Netherlands, have revealed, MRs play a role in behavioral reactivity during novel situations, whereas GRs are involved in the consolidation of learned information. Moreover, the neuroprotectant effect of naturally occurring corticosteroids is due to their binding at MRs, whereas synthetic corticosteroids, which selectively bind at GRs, render neurons susceptible to cell death. This does not negate the fact that continuous exposure to stress or acute serious trauma may have long-term effects, increasing an individual’s risk of developing stress-related diseases such as depression, anxiety, sleep disturbances, or post-traumatic stress disorder.
One possible explanation is the long-term effect induced by stress hormones on gene activity. It is of note that GRs and MRs, once activated by corticosteroids, act as powerful transcription factors that bind at specific DNA sequences to activate or repress transcription of a manifold of genes. Hormoneactivated MRs and GRs can also bind to other transcription factors and through this interaction indirectly modify the transcription rate of many genes that are not primarily hormone regulated. Stresselicited hormone secretion and its consequences on gene activity represents a perfect model for the study of the influence of environmental psychosocial factors on gene expression in the context of the pathophysiology of depression.
While it is well-established that the risk of developing depression depends on a complex multigenetic predisposition, it is an equally well-founded fact that depression can be precipitated by life events. On a functional level, such life events are associated with an increased demand to maintain homeostasis. This condition is associated with increases in stress hormone levels, which exert activity at the level of gene expression by transforming GRs and MRs into transcription factors. Under these demanding conditions subtle changes in the genetic blueprint, e.g., single nucleotide polymorphisms, may no longer be compensated, resulting in the emergence of a clinical phenotype. Given that stress hormones account for such effects by transducing environmental challenges on the cellular and genomic level, it would seem worthwhile to consider the reuptake options available for intervention. To better understand the rationale for today’s pharmacological strategies, the relevant facts about stress hormones are listed in Table 1. It is important to note that elevations of stress hormones or aberrant responses to neuroendocrine probes are not a byproduct of emotional stress associated with depression. As illustrated in Fig. 1, stress hormone abnormalities precede the occurrence of depressive psychopathology, and conversely, a gradual normalization of stress hormone regulation is followed by an improvement in depressive symptoms. This time-course pattern rejects the possibility that abnormal test results are secondary to depression. A more straightforward interpretation links regulation of the stress hormone system to the pathophysiology of depression (Holsboer 1995).
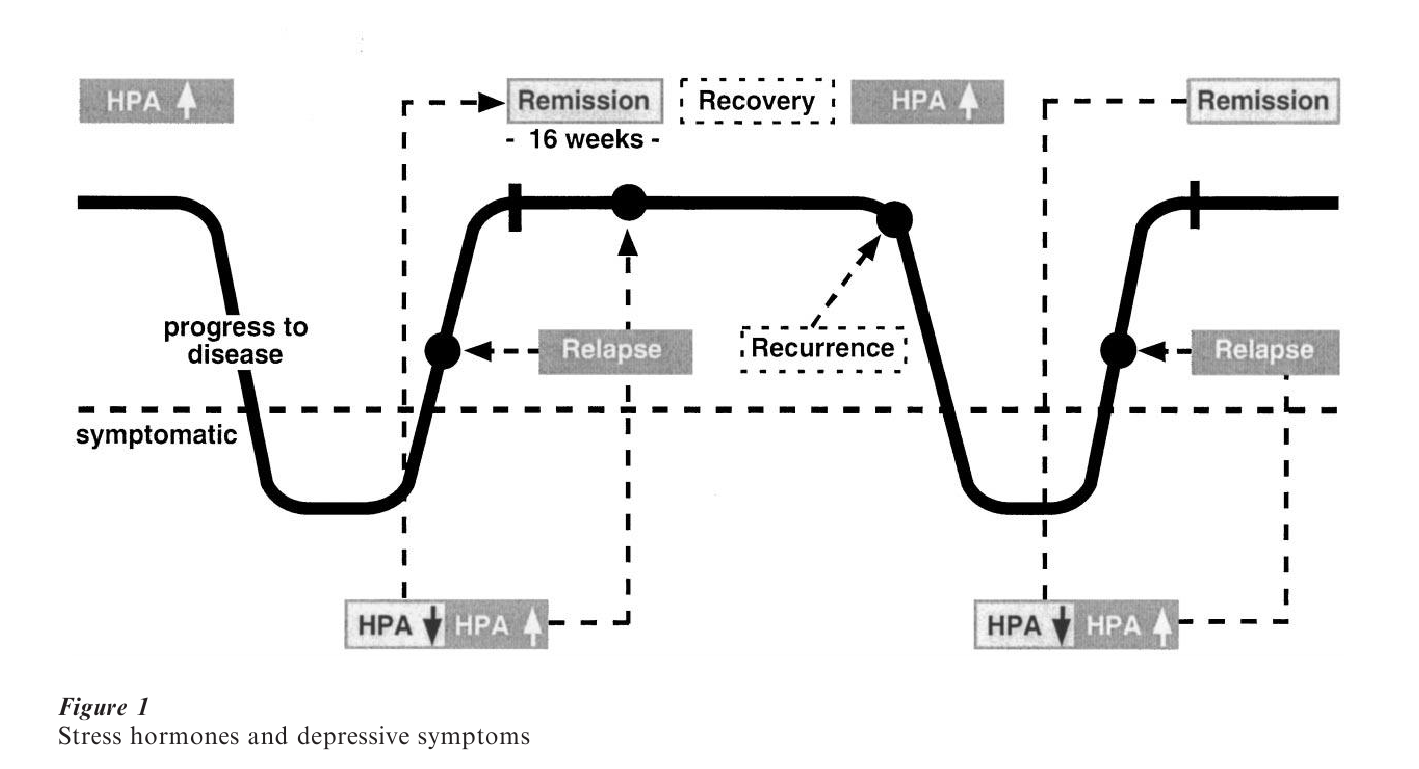
The key regulator of peripheral stress hormones is corticotropin-releasing hormone (CRH), a neuropeptide synthesized mainly in the nucleus paraventricularis of the hypothalamus, an assembly of brain nuclei that serves as a relay between brain function and peripheral autonomous and endocrine regulation (Holsboer 2000). From the hypothalamus, CRH is released into small blood vessels that allow transportation of CRH to the pituitary where it evokes the synthesis and release of corticotropin. This hormone enters the blood circulation and once it arrives at the adrenal cortex, cortisol is secreted, which regulates CRH and corticotropin release through a well-balanced feedback loop. Key elements in this circuit are corticosteroid receptors that when hormone-activated, serve as a break on genes coding for CRH and corticotropin. The corticosteroid receptor hypothesis of depression submits that the signaling through these receptor molecules is impaired, resulting in a number of changes of gene activity, including that coding for CRH. This neuropeptide is not only involved in organizing the stress hormone system—which is traditionally called the ‘hypothalamic-pituitary-adrenocortical (HPA) axis, although it involves many other brain areas including those believed to be involved in the neuropathology of depression—but also coordinates a number of behavioral signs and symptoms that aid for adaptation to stress.

In transgenic mice continuous overexpression of CRH, as documented to occur in depression (see Table 1), leads to increased anxiety-like behavior and other symptoms reminiscent of depression (decreased appetite, sleep disturbance, loss of libido, etc.) (Steckler et al. 1999). Similar findings in rats to which CRH was administered also suggest that CRH precipitates behavioral changes compatible with anxiety and the symptoms of depression. Consequently, research has focused on the possibility of reducing these negative effects of CRH by blocking its receptors. After the responsible receptor had been identified as the CRH-R1 subtype, drugs were developed that are capable of antagonizing the actions of CRH specifically at these receptors. Indeed, in animal studies it has been shown that anxiety-like behavior under stress conditions or under conditions of withdrawal from drug abuse disappears when CRH-R1 antagonists are administered. Moreover, stress-induced sleep disturbances disappeared in rats when the administration of the stressor is combined with the CRH-R1 antagonist. On a neurochemical level it has been shown that CRH overproduction in the brain is associated with blunted serotonin responses to stress, whereas genetic invalidation of CRH-R1 receptors results in increased serotonin release. This neurotransmitter is strongly implicated in the pathophysiology of depression, and most currently prescribed antidepressants such as fluoxetine, paroxetine, and citalopram work on the principle of the enhancement of serotonin release. This favors the hypothesis that blockers of CRH type 1 receptors may also work as antidepressants through the enhancement of serotonergic neurotransmission and the correction of excessive central stress hormone drive. These considerations have led to the first clinical trial of a CRH-R1 antagonist in depression and its results have confirmed that this hypothesis-driven research has led to a novel pharmacotherapeutic concept (Holsboer 1999).
2. Sex Hormones
The differences between men and women extend to neuroanatomy, with male brains having a higher volume of cerebrospinal fluid and grey matter. Women’s brains are denser in grey matter, consisting of neuronal cell tissue and connection-making dendrites. It is believed that the proportional difference between grey and white matter accounts for the different performances in neuropsychological tests where men demonstrate better spatial skills, but women are more verbally proficient. Moreover, estrogens, women’s key sex hormone, secreted from gonadal glands, may explain some of the functional gender differences. These can be studied in postmenopausal women, whose endogenous estrogen production is reduced. Studies using a brain imaging technique have shown that estrogen administration to postmenopausal women can activate a brain region responsible for storing phonologic information, which may explain why women score higher in language tests.
Changes in estrogen secretion have been related to many psychiatric syndromes ranging from schizophrenia to depression and Alzheimer’s disease. While the lifetime risk for schizophrenia is equal for men and women, it has been found that the age at which affected men have their first psychotic symptoms is three to four years earlier than in women who, after the menopause, have a second peak of disease onset. The later onset of schizophrenia in women has led to the hypothesis that estrogens protract the expression of schizophrenic symptoms (Hafner et al. 1993). One possible mechanism involved is an estrogen-induced decrease in dopaminergic neurotransmission. This attractive hypothesis still awaits corroberation from therapeutic tests using estrogens in combination with neuroleptics. Further studies will also have to address the contradiction of estrogen-elicited increases in the density of serotonin type 2A receptors in the forebrain and the role of these receptors in the pathogenesis, as well as in the treatment of psychoses. Numerous studies have shown that serotonin type 2A receptor blockade is a fundamental pharmacological requirement for the new drugs termed atypical antipsychotics, such as clozapine or olanzapine. On the other hand, serotonin type 2A receptor density has been found to be diminished in patients with schizophrenia, which would be consonant with decreased estrogen secretion with age.
Estrogens are widely prescribed, mainly by gynecologists and practitioners to alleviate mood swings associated with the premenstrual syndrome (Schmidt et al. 1998). Comparatively little systematic research has been conducted, although most published trials have reported beneficial effects. Whether estrogens are also effective in major depression is controversial, and careful inspection of the available data casts doubt on the possibility that the documented effects of estrogens upon serotonergic (increase of serotonergic 2A receptors in the forebrain), noradrenergic, dopaminergic, and cholinergic systems may translate into a psychotropic action comparable to established antidepressant drugs. The situation is perhaps different in older women where endogenous estrogen production is reduced, or in women where massive changes in estrogen secretion have occurred. Postpartal depression is a typical example of this condition and in these cases estrogen treatment has been reported to be beneficial. Perhaps the most meaningful application of estrogens in women with depression is its use as an adjunct to standard antidepressant treatment. For example, estrogen replacement treatment in combination with fluoxetine was found to be superior to antidepressant monotherapy in women who were 60 years or older. Another study where postmenopausal women with severe major depression were treated with imipramine, and where to those who did not respond either to estradiol or placebo was added as adjunct, the investigators failed to observe an improvement by the hormone. In the light of this, mild dysthymic and depressive symptoms may respond to estrogen supplementation either directly through central neuropharmacological actions or indirectly through the beneficial effects of estrogen on peripheral bodily functions such as libido, hot flushes, skin crawls, rheumatic pains, etc. (Santoro et al. 1999).
Perhaps the most compelling influence of estrogen on brain function is related to memory, cognition, and the apparent protective effect of estrogens with respect to the onset of Alzheimer’s dementia. Multiple epidemiological studies have suggested that estrogen replacement therapy may protect against, delay the onset of, or slow the cognitive decline associated with Alzheimer’s disease. In these studies, women who took estrogen replacement once they reached menopause had a significantly decreased likelihood of developing Alzheimer’s disease. A cautionary note, however, seems warranted as this correlation does not necessarily reflect a causal mechanism. In favor of a specific role of estrogens as neuroprotectants are the results of a number of experiments investigating the interaction of these hormones with β-amyloid, which is thought to be causal in the development of cerebral plaques. These are in combination with fibrillary tangles, a characteristic neuropathologic feature of Alzheimer’s disease. Estradial was found to protect against βamyloid-induced cell degeneration and to reduce the production of this β-amyloid. The protective effect does not seem to involve estrogen receptor activation but rather is related to the antioxidative actions of these steroid hormones (Behl and Holsboer 1999). This is a clinically pertinent observation because the decreased risk of developing Alzheimer’s disease might be outweighed by the increased risk of, for example, breast cancer (Grodstein et al. 1997). It was shown that the antioxidant effect of estrogens is independent of the hormonal effects via estrogen receptors. This opens up the possibility of developing ‘designer estrogens’ to provide long-term protection against the common causes of morbidity and mortality such as osteoporosis, coronary heart disease, and probably against cognitive decline associated with Alzheimer’s disease.
Male sex hormones have only received scant attention in psychiatry despite their widespread use as anabolic drugs. The best-established effects of male sex hormones, especially of testosterone, are related to their effect on sexual function. Sexual desire, intense sexual feelings, and sexual activity are reduced in males with pathologically decreased testosterone levels, and are restored with testosterone treatment. In young women without sexual dysfunction one single dose of testosterone can induce markedly increased sexual arousal irrespective of sex hormone secretory activity. However, men who are not hypogonadal, i.e., have normal plasma testosterone levels, but a decreased libido and erectile function, do not respond to testosterone administration. In other words, decreased sexual arousal and erectile impotence are not responsive to testosterone in men with normal sex hormone status. That male sex hormones play a role in aggression has been inferred from studies in prisons, in which violent criminals were compared with nonviolent inmates. Many, but not all, of these studies agree that violent criminals have higher testosterone concentrations in both plasma and cerebrospinal fluid.
Perhaps the more important question relates to the psychiatric risk of testosterone administration in dosages used by young men to improve athletic performance or personal appearance (‘body builders’). A recent examination of the effects of testosterone on mood and aggressive behavior found notable adverse psychiatric effects in a subgroup of 16 percent of the study population, which was found to exhibit manic symptoms. From animal experiments a large number of interactions of testosterone with neurotransmitters and neuropeptides have emerged. These findings, however, give no hint as to why supraphysiological dosages of testosterone might induce manic symptoms in a fraction of otherwise healthy males. There is no documented evidence that patients with mania have elevated testosterone despite their increased activity levels, including sexual activity. Likewise, no data exist showing that testosterone is decreased in depression and that men with plasma testosterone levels within the normal range benefit from testosterone treatment.
What still needs to be investigated is whether male sex hormones are beneficial as neuroprotectants and, specifically, if they are beneficial in reducing the formation of β-amyloid, the protein related to Alzheimer’s disease. It is of note that the brain contains an enzyme that converts testosterone into estrogen.
3. Neuroactive Steroids—Dehydroepiandrosterone
A special situation has developed with regard to dehydroepiandrosterone (DHEA), which has received disproportional media coverage and has been promoted as a ‘fountain of youth.’ This steroid was first described by Adolf Butenandt, who pioneered steroid biochemistry and who later received the Nobel prize in recognition of his work. DHEA is often thought to be a ‘weak androgen,’ which is incorrect as it is produced only to a small extent by gonadal glands and stems mainly from the adrenal cortex. The error possibly derives from the fact that testosterone and DHEA decrease with increasing age, whereas cortisol secretion increases. The increasing cortisol: DHEA ratio with age, however, reflects the fact that the part of the adrenal cortex from which cortisol is secreted (zona fasciculata) remains unchanged, while the part of the adrenal cortex from where DHEA is produced (zona reticularis) gradually undergoes apoptotic change with the consequence that the very high level of DHEA, which is present mainly as sulfate conjugate (DHEAS), declines (Arlt et al. 1999). Particularly low DHEAS levels have been recorded in patients under chronic stress or with serious health problems ranging from arthritis to HIV infection.
DHEAS is produced not only in the adrenal cortex, but also in the brain. Indeed, plasma DHEA concentrations in rats have been found to be lower than those in the brain and this is not due to the accumulation of peripherally produced steroid. In contrast, astrocytes and neurons are capable of synthesizing DHEA from pregnenolone, a common steroid precursor molecule and a derivative of cholesterol. Because nerve cells have the capacity to produce DHEA, this steroid is called a neurosteroid (Rupprecht and Holsboer 1999). The main effect of DHEA in the brain is its antagonistic effect on gamma-aminobutyric acid A (GABAA) receptors. Based on this pharmacological property, several studies in animals and humans were initiated that observed the memory-enhancing effects of DHEA, compatible with the actions of other GABAA antagonists. The finding of a DHEA-induced increase in rapid eye movement sleep, which has been implicated in memory storage, is in accord with this notion. In the USA DHEA has become an ‘over-the counter’ remedy for all ailments associated with aging, but this occurs in the absence of a solid database that would allow the use of DHEA in clinical treatment.
Another recent approach is an initiative in France where 280 men and women between 60 and 79 years received 50 mg DHEA or placebo daily for one year (Baulieu et al. 2000). The overall clinical impression obtained from the data of these healthy individuals was that this kind of replacement therapy has beneficial effects on bone and skin status as well as on several behavioral characteristics including sexual function. More refined analysis of this study, when available, will perhaps help us to identify the role of DHEA in preventive medicine. Until such data are made accessible to the public, DHEA will be considered by health care professionals as a ‘lifestyle’ drug. The only exception that relates to psychiatric conditions are studies where DHEA was administered to patients with depression where DHEA levels are low. Preliminary results from these studies noted an improvement in ratings of depression and aspects of memory function, supporting the notion that decreased DHEA levels are amenable to pharmacotherapy in selected cases.
4. Thyroid Hormones
There is a well-documented relationship between thyroid disease and psychiatric symptomatology. In patients with hypothyroidism, depressive symptoms are very frequent and disappear when thyroxine (T4) or triiodothyronine (T3) is given as substitution. These two hormones are secreted by the thyroid gland and act at the interior pituitary to inhibit thyroid-stimulating hormone (TSH), and at the hypothalamus to inhibit the synthesis and secretion of thyrotropinreleasing hormone (TRH).
At baseline, patients with depression are euthymic, i.e., they have normal T3, T4 and TSH levels. However, when the pituitary is stimulated with TRH, about 25 percent of the patients show blunted TSH responses, which correlates with the CRH-elicited corticotropin increases. The blunted TSH response is explained by chronic hypersecretion of TRH from the hypothalamus producing desensitization of anterior pituitary TRH receptors and ultimately reducing responsiveness to exogenous TRH. These laboratory findings do not suggest that hypothyroidism should be treated with thyroid hormones, which, nevertheless, have been used in various ways to treat depressive illness, mainly in combination with other antidepressants. In the treatment of depression, T3 mainly is used because this hormone is the active steroid hormone in the brain, locally converted from T4 by a brain-specific enzyme called deiodinase that removes one iodine atom from the thyroxine hormone molecule.
The finding that brain concentrations of T3 are as high as those of T4, whereas serum T4 levels are 50 times higher than those of T3 is a strong argument for a key role of T3 in the brain. T3 formation is increased by antidepressants that increase the aforementioned deiodinase enzyme activity. T enters neurons readily and binds to nuclear receptors modulating the activity of many genes directly or through protein-protein interactions. As of yet it is entirely unclear which of T3’s neurochemical effects are involved in its beneficial effects on brain function. It is of note, however, that T3 affects the serotonergic system as, for example, T3 administration to rats increases basal serotonin levels in the forebrain. Similarly, T3 has been found to induce subsensitivity of presynaptic serotonin 1A autoreceptors, proposed as one of the major mechanisms responsible for the antidepressant effect of selective serotinin reuptake inhibitors such as fluoxetine. In line with the special role of T3 in brain function is the finding that psychopathological and neuropsychological symptoms of hypothyroidism respond favorably, if the patients receive a balanced combination of T3 and T4 instead of the usual T4 monotherapy.
T3 has been used in doses of 25 µg/day in combination with antidepressants and most reports claimed that adjunctive thyroid hormone treatment may hasten the onset of response (Newman et al. 2000). While most of these studies reported intriguing findings, they are preliminary and await confirmation in studies using current standard methods for efficacy validation. Another interesting approach using T3 is the augmentation of the antidepressant effect in patients who have failed to respond to their current antidepressant therapy. Nonresponse, partial response, or failure to achieve stable remission frequently haunt practicing clinicians. Many studies exist reporting that T3 can amplify the antidepressant effect thereby shifting nonresponders or partial responders into responders. A meta-analysis found that all T3 augmentation studies could confirm that T3 was more effective than placebo when combined with antidepressants. However, when more rigid standards were applied, e.g., randomized control design, etc., the significant difference between T3 and placebo as potential augmentors of antidepressant action disappeared. Comparing all available augmentation strategies, it seems clear that T3 studies have provided the most convincing database and that this justifies a critical evaluation of T3 dosage and duration of treatment in combination with new antidepressants.
5. Summary And Outlook
This research paper has taken a few examples of how neuroendocrinology has shaped psychiatric research, although hormone treatments have not yet entered clinical routine. Estrogen replacement therapy is wellestablished, but it is doubtful that it works as an antidepressant and its clinical usefulness in preventing Alzheimer’s disease is not yet validated. Whether similar effects can be expected from testosterone replacement in men is an open question. DHEA treatment has received much attention, but its promise as a rejuvenating hormone still has to stand the test of time. Although not unanimously accepted, the most robust database exists for T augmentation studies, which need to be corroborated by applying current validation standards for clinical trials. The most exciting development to date is the novel opportunity to interfere with drugs that block the anxiogenic and depression-like effects of CRH. Perhaps this new pharmacology can put hormone treatment back on the map in psychopharmacology.
Where will the future lead us? Classically, a neuroendocrine factor has been defined based on phenotypic changes. Entering the era of functional genomics, we will depart from studying one gene at a time and will rather apply a global approach, which will result in a dramatic expansion of our knowledge of gene sequences, expression profiles, and protein functions. From this rapidly progressing genomic revolution we can expect the discovery of many new genes in the endocrine systems. This will pave the way to the identification of new drug targets and the analysis of the genetic susceptibility of individuals to develop diseases where endocrine dysfunction, e.g., impaired stress hormone regulation, plays a role. Finally, if single nucleotide polymorphisms are mapped for gene areas of interest, customized therapies for the prevention of singularly detrimental effects of hormone dysregulation will become available. These new opportunities will establish a new platform where endocrinology works in partnership with psychiatry at a behavioral, functional, and genomic level.
Bibliography:
- Almeida O F, Conde G L, Crochemore C, Demeneix B A, Fischer D, Hassan A H S, Meyer M, Holsboer F, Michaelidis T M 2000 Subtle shifts in the ratio between pro and antiapoptotic molecules after activation of corticosteroid receptors decide neuronal fate. FASEB Journal 14: 779–90
- Arlt W, Haas J, Callies F, Reincke M, Hubler D, Oettel M, Ernst M, Schulte H M, Allolio B 1999 Biotransformation of oral dehydroepiandrosterone in elderly men: Significant increase in circulating estrogens. Journal of Clinical Endocrinology and Metabolism 84: 2170–6
- Baulieu E-E, Thomas G, Legrain S, Lahlou N, Roger M, Debuire B, Faucounau V, Girard L, Hervy M P, Latour F, Leaud M C, Mokrane A, Pitti-Ferrandi H, Trivalle C, de Lacharriere O, Nouveau S, Rakoto-Arison B, Souberbielle JC, Raison J, Le Bouc Y, Raynaud A, Girerd X, Forette F 2000 Dehydroepiandrosterone (DHEA), DHEA sulfate, and aging: Contribution of the DHEAge Study to a sociobiomedical issue. Proceedings of the National Academy of Science of the United States of America 97: 4279–84
- Behl C, Holsboer F 1999 The female sex hormone oestrogen as neuroprotectant. Trends in Pharmacological Sciences 20: 441–4
- de Kloet E R, Oitzl M S, Joels M 1999 Stress and cognition: Are corticosteroids good or bad guys? Trends in Neurosciences 22: 422–6
- Grodstein F, Stampfer M J, Colditz G A, Willett W C, Manson J E, Joffe M, Rosner B, Fuchs C, Hankinson S E, Hunter D J, Hennekens C H, Speizer F E 1997 Postmenopausal hormone therapy and mortality. New England Journal of Medicine 336: 1769–75
- Hafner H, Riecher-Rossler A, an der Heiden W, Mauer K, Fatkenheuer B, Loffler W 1993 Generating and testing a causal explanation of the gender difference in age at first onset of schizophrenia. Psychological Medicine 23: 925–40
- Holsboer F 1995 Neuroendocrinology of mood disorders. In: Bloom F E, Kupfer D (eds.) Psychopharmacology: The Fourth Generation of Progress. Raven Press, New York, pp. 957–69
- Holsboer F 1999 The rationale for corticotropin-releasing hormone receptor (CRH-R) antagonists to treat depression and anxiety. Journal of Psychiatric Research 33: 181–214
- Holsboer F 2000 The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology 23: 477–501
- Newman M E, Agid O, Gur E, Lerer B 2000 Pharmacological mechanisms of T3 augmentation of antidepressant action. International Journal of Neuropsychopharmacology 3: 187–91
- Rupprecht R, Holsboer F 1999 Neuroactive steroids: Mechanisms of action and neuropsychopharmacological perspectives. Trends in Neurosciences 22: 410–16
- Santoro N F, Col N F, Eckman M H, Wong J B, Pauker S G, Cauley J A, Zmuda J, Crawford S, Johannes C B, Rossouw J E, Merz N B 1999 Therapeutic controversy. Hormone replacement therapy—where are we going? Journal of Clinical Endocrinology and Metabolism 84: 1798–1812
- Schmidt P J, Nieman L K, Danaceau M A, Adams L F, Rubinow D R 1998 Differential behavioral effects of gonadal steroids in women with and in those without premenstrual syndrome. New England Journal of Medicine 338: 209–16
- Steckler T, Holsboer F, Reul J M H M 1999 Glucocorticoids and depression. Best Practice & Research Clinical Endocrinology and Metabolism 13: 597–614
ORDER HIGH QUALITY CUSTOM PAPER

Always on-time

Plagiarism-Free

100% Confidentiality