
View sample sleep and biological clocks research paper. Browse research paper examples for more inspiration. If you need a psychology research paper written according to all the academic standards, you can always turn to our experienced writers for help. This is how your paper can get an A! Feel free to contact our writing service for professional assistance. We offer high-quality assignments for reasonable rates.
One of the most remarkable characteristics of life on earth is the ability of almost all species to change their behavior on a 24-hour basis. Not only are daily rhythms observed in organisms as diverse as algae, fruit flies, rodents, and humans, but every aspect of the internal environment of the organism also undergoes pronounced fluctuations over the course of the 24-hour day. These fluctuations or oscillations are called circadian rhythms. Circadian, from the Latin circa diem, means approximately one day. An oscillation is defined by its period (i.e., the time interval after which the wave shape of the oscillation recurs), by its range (i.e., the difference between the maximal and minimal values within one period), and by its mean value (i.e., the arithmetic mean of all instantaneous values of the oscillating variable within one period). Half the range of oscillation is called the amplitude. The peak value is often referred to as the acrophase, and the lowest value is often called the nadir of the oscillation.
Academic Writing, Editing, Proofreading, And Problem Solving Services
Get 10% OFF with 24START discount code
An immense variety of circadian rhythms have been observed in humans—from physiological variables such as body temperature, heart rate, and blood pressure to behavioral variables such as mood, vigilance, and cognitive performance. The temporal organization of behavioral and physiological variables across the 24-hour cycle ultimately results from the activity of two interacting time-keeping mechanisms in the central nervous system: endogenous circadian rhythmicity and sleep-wake homeostasis. Depending on the parameter under consideration, the wave shape of the rhythm may also be affected by food intake, postural changes, changes in intensity of physical activity, and effects of stress. In mammals, endogenous circadian rhythmicity is generated by a pacemaker located in the paired suprachiasmatic nucleus (SCN) of the hypothalamus (Turek, 1998). Sleep-wake homeostasis refers to an hourglass-like mechanism relating the amount and quality of sleep to the duration of prior wakefulness (Borbély, 1998). Both processes interact to control temporal changes in essentially all behavioral and physiological variables across the 24-hour day, but the relative contributions of each of these factors vary from one variable to the other.
In the following three sections, we review current notions on the human circadian system, sleep regulation, and the interaction of circadian rhythmicity and sleep in the control of the 24-hour profiles of behavioral and physiological variables under normal conditions. The last section is dedicated to abnormal conditions of circadian rhythmicity, sleep, or both, either behaviorally induced or resulting from pathological alterations.
Circadian Rhythmicity
Overview
Circadian rhythms are endogenous—that is, they originate from within the organism and persist even under constant environmental conditions. The endogenous nature of human circadian rhythms has been established by experiments in which subjects were isolated with no access to the natural light-dark (LD) cycle and no time cues. Such experiments were first performed in natural caves, then in underground bunker laboratories, and finally in specially designed windowless soundproof chambers. The results of such experiments showed that circadian rhythms continue to be expressed even in the absence of external time-giving cues. However, under such constant environmental conditions, the period of the rhythm in humans has been commonly observed to be slightly longer than 24 hours. When a circadian rhythm is expressed in the absence of any 24-hour signals in the environment, it is said to be free-running (i.e., the rhythm is not synchronized or entrained by any cyclic change in the physical environment). The period length of a free-running rhythm varies between individuals.
The fact that the endogenous circadian period observed under constant conditions is not exactly equal to 24 hours implies that periodic changes in the physical environment must synchronize or entrain the internal-endogenous clock system regulating circadian rhythms. Aclock with a period even only a few minutes shorter or longer than 24 hours would otherwise be soon out of synchrony with the environmental day. Factors that are capable of entraining or synchronizing circadian rhythms are called zeitgebers, a German word for time giver. For the vast majority of mammalian species, the LD cycle is the most powerful environmental factor that synchronizes the endogenous biological clock, but entrainment by other periodic factors in the environment (e.g., social cues, feeding schedules) has been demonstrated. In rodents and other mammals, social and behavioral cues that alter the restactivity cycle—either by eliciting activity during the normal rest period or by preventing activity during the normal active period—result in phase shifts of circadian rhythms. In humans, there is evidence to indicate that exposure to dark or sleep during the usual active period and exposure to high levels of physical activity during the usual rest period can phaseshift circadian rhythms.
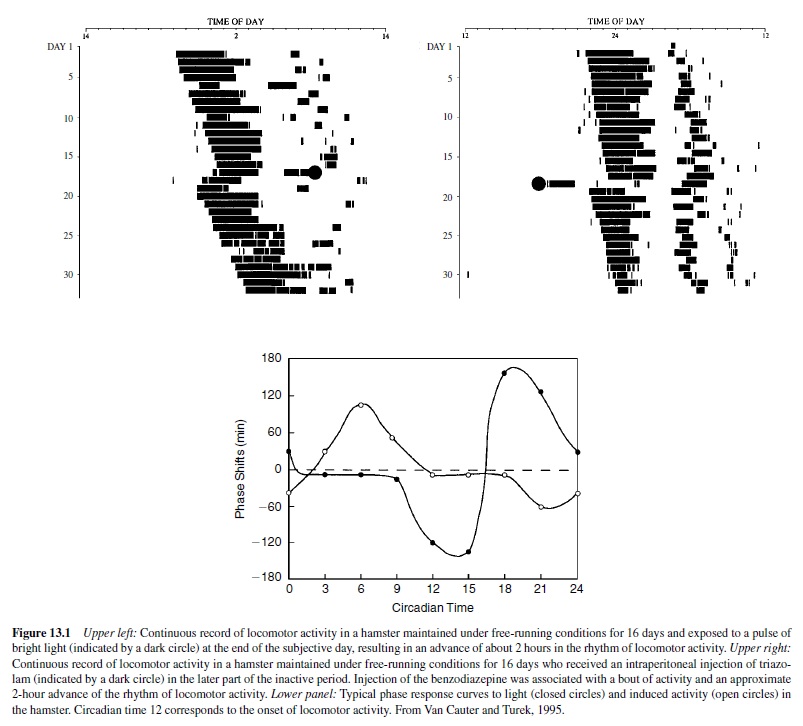
The direction and magnitude of the shifts resulting from exposure to a zeitgeber depend on the circadian time at which the zeitgeber is presented. Aplot of the phase shift induced by an environmental perturbation as a function of the circadian time at which the perturbation is given is called a phase response curve (PRC). The upper left panel of Figure 13.1 shows a typical response of the rhythm of locomotor activity to a pulse of bright light in a hamster. The animal is kept in constant darkness for 16 days and the rhythm of locomotor activity free-runs. A pulse of light presented at the end of the 17th subjective day results in a phase advance of the onset of locomotor activity. The lower panel of Figure 13.1 shows a typical PRC to pulses of bright light (closed symbols) in the hamster derived from experiments such as that illustrated in the upper left panel. The PRC to light pulses for all organisms share certain characteristics including the fact that light pulses presented near the onset of the subjective night induce phase delays, whereas light pulses presented in the late subjective night or early subjective day induce phase advances. Light pulses presented during most of the subjective day induce no phase shifts. The upper right panel of Figure 13.1 shows the phase advance of the onset of locomotor activity when a bout of activity is pharmacologically induced during the normal rest period. The PRC to this nonphotic zeitgeber is shown (open symbols) in the lower panel of Figure 13.1 (Van Cauter & Turek, 1995).
Circadian rhythms have evolutionary importance and functional significance because they provide synchronization with the pronounced periodic fluctuations in the external environment and organize the internal milieu so that there is coordination and synchronization of internal processes. The external synchronization is of obvious importance for the survival of the species and ensures that the organism does the right thing at the right time of the day. The internal synchronization ensures temporal organization between the myriad of biochemical and physiological systems in the body. The physical and mental malaise occurring following rapid travel across time zones (i.e., jet lag) and the pathologies associated with longterm shift work are assumed to be due in part to the misalignment between various internal rhythms.
The Organization of the Mammalian Circadian System
As previously noted, the endogenous component of mammalian 24-hour rhythms originates from a pacemaker located in the paired SCN of the anterior hypothalamus (Turek, 1998). The two halves of the SCN each contain about 8,000 neurons, and it appears that many SCN neurons are capable of independent oscillatory behavior (Dunlap, 2000). There is strong evidence to indicate that the SCN is the master pacemaker involved in the generation and entrainment of circadian rhythms. Indeed, when the SCN is destroyed in rodents, circadian rhythmicity is abolished or markedly disrupted. Circadian rhythmicity can be restored in adult arrhythmic SCN-lesioned rodents by transplanting fetal SCN tissue into the region of the SCN. Moreover, if tissue from a fetus with a mutant clock (i.e., an animal that when adult shows an abnormal circadian rhythm) is used, the host animal will show the rhythm of the donor genotype (Ralph, Foster, Davis, & Menaker, 1990). Finally, a number of SCN rhythms persist in vitro, including those of neural firing, vasopressin release, and glucose metabolism.
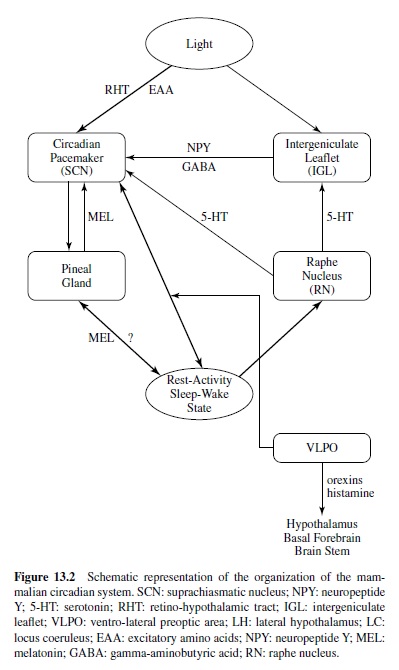
Figure 13.2 provides a schematic representation of the current understanding of the organization of the mammalian circadian system (Van Cauter & Turek, 2001). The SCN receives light information from the retina via the retinohypothalamic tract (RHT) using glutamatergic neurotransmission. In addition to the RHT, the SCN also receives retinal information indirectly from the intergeniculate leaflet (IGL) of the thalamus, which receives direct light input from the retina. Transmission from the IGL to the SCN involves both neuropeptide Y (NPY) and gamma-aminobutyric acid (GABA) inputs. Photic information received by the SCN is transmitted to the pineal gland, which does not receive any direct photic input in mammals. The pineal gland releases the hormone melatonin, which is secreted during the dark period only and has sedative properties. Some evidence indicates that melatonin exerts phase-setting effects on circadian function, and melatonin receptors have been identified in the SCN. The SCN also receives a direct serotoninergic (5-HT [5-hydroxytryptamine]) input from the raphe nucleus (RN). The median and dorsal raphe nuclei send robust 5-HT projections to the ventrolateral SCN and the IGL. Recent studies indicate that photic information can influence the effects of 5-HT stimulation on circadian rhythmicity, and 5-HT inputs can alter the response of the clock to the entraining effects of light (Turek & Van Reeth, 1996). One of the pathways by which 5-HT information may mediate the effects of activity-inducing stimuli on the circadian clock may involve the NPY projection from the IGL to the SCN. Thus, the IGLis currently seen as a putative integrator of photic and nonphotic information in the circadian system.
Circadian control of the sleep-wake cycle is likely to involve several pathways. There is a prominent indirect projection from the SCN to the locus coeruleus (LC), a noradrenergic center that plays a major role in the control of arousal (Aston-Jones, Chen, Zhu, & Oshinsky, 2001). The ventrolateral preoptic (VLPO) area of the hypothalamus contains sleep-active neurons, which provide GABAergic innervation to inhibit the ascending arousal monoamine systems, including the LC (Gallopin et al., 2000; Sherin, Shiromani, McCarley, & Saper, 1996). There is a circadian rhythm in c-fos expression in the VLPO, suggesting that this area could also be involved in the circadian control of sleep and wake states. Recent work also suggests that the SCN may modulate activity in an area of the lateral hypothalamus where neurons containing the neuropeptide orexin appear to participate in the regulation of sleep and wakefulness.
The past decade has seen a great deal of progress in the understanding of the molecular and genetic mechanisms underlying circadian rhythmicity. The first mammalian clock gene, Clock, was identified and cloned in the mouse. The identification of the other known mammalian circadian genes (bmal, tim, cry1, cry2, per1, per2, per3), and the study of the interactions of these genes and their protein products, led to the elucidation of a transcription-translation autoregulatory loop (King & Takahashi, 2000). This complex, negative feedback loop generates a remarkably precise oscillation of about 24-hour CLOCK and BMAL proteins bind together to activate expression of the per1, per2, per3 genes. The PER proteins form dimers with other proteins (PER, TIM, CRY1, CRY2), reenter the nucleus, and in turn suppress the CLOCK and BMAL complex, eventually halting per gene production. As PER protein levels decline, the inhibition of CLOCK and BMAL is released and the loop begins anew. A recently discovered mammalian circadian gene, casein kinase I epsilon (Lowrey et al., 2000), modulates the timing of this feedback loop by phosphorylation and degradation of PER protein.
The plant photopigment-like molecules from the cryptochromes GENES (cry1 and cry2) also appear to play a role in the light input pathway to the SCN (Sancar, 2000). The mechanisms by which the central circadian pacemaker drives downstream effector systems are poorly understood.
Ontogeny of Circadian Rhythms
The SCN is formed in the rat between embryonic day 14 (E14) and E17 (approximately 5 days before birth; Davis, Frank, & Heller, 1999; Turek & Van Reeth, 1996). Between E17 and postnatal day 10 (P10), the SCN enlarges and assumes its adult appearance. Synaptogenesis occurs primarily after birth, because it has been estimated that each adult SCN neuron has 300–1,200 synaptic contacts, and at E19 there appears to be less than one synapse per SCN neuron. This is particularly interesting in view of the fact that rhythmicity within the SCN is present before this time. In addition, the innervation of the SCN by the RHT is a postnatal event and becomes clearly present by P4.
In several mammalian species, circadian oscillations are present in the SCN already during fetal life (Reppert & Weaver, 1991). The fetal SCN is entrained by circadian signals from the mother, and although rhythms develop normally in pups born to SCN-lesioned mothers, fetal SCN rhythms cannot be entrained without the presence of the maternal SCN (Reppert & Weaver, 1991). It seems likely that the fetal SCN is entrained by multiple circadian signals from the mother, and at least in rats, the maternal influence on rhythmicity persists after birth for about a week. After a week, the pups become capable of responding to the entraining effects of the light-dark cycle.
Despite the early development of circadian rhythms within the SCN, the ability of the SCN to regulate overt behavioral and physiological rhythms does not occur until much later. For example, in rats, most endocrine and behavioral rhythms do not appear until the second or third weeks of life. Therefore, the circadian clock in the SCN becomes a circadian pacemaker when it develops sufficient afferent, intrinsic, and efferent neural connections to be entrained and to regulate effector systems (Moore, 1991). However, it has been demonstrated that the phase of specific rhythms can be set long before the appearance of the rhythms (Davis et al., 1999; Reppert & Weaver, 1991; Turek & Van Reeth, 1996).
Moore (1991) divided the development of circadian function regulated by the SCN into four components. First, the cells in the SCN are formed, they grow and mature, and rhythmic function is established within the nucleus before major synaptic contacts either between SCN cells or with the rest of the brain are formed. Second, there is the development of entrainment pathways to the SCN (that is primarily the RHT) with the resultant ability of the SCN to respond to environmental cues. Third, SCN projections develop, resulting in the coupling of the SCN to effector systems, often before the effector systems are able to express rhythms. Fourth, the maturation of the output systems reaches the point at which they can express circadian function.
Aging of the Circadian System
Age-related changes have been documented in metabolic, endocrine, and behavioral circadian rhythms in a variety of species, including humans (Brock, 1991; Carrier, Monk, Buysse, & Kupfer, 1996; Copinschi, Leproult, & Van Cauter, 2001; Czeisler, Chiasera, & Duffy, 1991; Kolker & Turek, 2001; McGinty & Stern, 1988). Among the observed age-related alterations are reduced amplitude, increased day-to-day variability, changes in intrinsic endogenous period, changes in the phase angle of entrainment, and diminished responses to synchronizing factors, including the light-dark cycle. The nature and severity of age-related disturbances appear to vary from one overt rhythm to another and there seem to be important species differences, in addition to the individual variability that is typical of age effects. The most frequently observed age-related alteration is a reduction in amplitude.
Changes in the waveshape or amplitude of a given circadian rhythm do not necessarily imply a change that is intrinsic to the circadian clock system itself. These changes could be explained by age-related changes that are either upstream (inputs to the pacemaker system) or downstream (outputs from the pacemaker system) from the circadian clock. For example, age-related changes in amplitude or entrainment of circadian rhythms can be caused by a decrease in the perception of light (as occurs in many older adults) or can reflect an alteration in the function of the effector system, such as a reduced ability to sustain locomotor activity, as has been observed in older laboratory rodents.
Nevertheless, there is evidence in rodents that the circadian pacemaker system itself is altered in aging.Anumber of studies have demonstrated that the free-running period of the circadian rhythms of locomotor activity, body temperature, or bothbecomesshorterwithageinrats,fieldmice,andhamsters (reviewed in Kolker & Turek, 2001). It is possible that the shortening of the intrinsic period of the circadian clock with age underlies the advance of circadian phase that is observed under entrained conditions in a number of species, including humans. Evidence for age-related changes in the clock itself has also been provided by Wise and her colleagues (Wise, Cohen, Weiland, & London, 1988), who found that aging alters the circadian rhythms of glucose utilization in the SCN. In both rats and hamsters, there are age differences in the amplitude of the firing rhythms of the SCN (Satinoff et al., 1993), but the number of neurons within the SCN does not appear to decrease with advanced age. Such differences in amplitude of intrinsicSCNrhythmicityarebestexplainedbyanage-related alterations of the coupling of the individual neuronal oscillators within the SCN. In humans, the most recent estimations of the length of the endogenous circadian period have suggested that the period remains constant across adulthood, at least for individuals that are typical of successful aging (Czeisler et al., 1999). Therefore, the well-documented agerelated phase-advances and decreases in amplitude of human circadian rhythms may reflect a modification of entrainment mechanisms, function of effector systems, or both.
In addition to intrinsic changes that may take place in the circadian pacemaker with senescence, there are also alterations in the response of the pacemaker to environmental stimuli. Old hamsters, rats, and mice show a decreased response to the phase-shifting effects of light pulses (Kolker & Turek, 2001). Aging also appears to attenuate the phase shifts induced by activity-inducing stimuli (Mrosovsky, 1996; Penev, Zee, Wallen, & Turek, 1995; Van Reeth, Zhang, Reddy, Zee, & Turek, 1993;Van Reeth, Zhang, Zee, &Turek, 1992).
Behavioral changes in older adults may also lead to changes in environmental inputs to the biological pacemaker system. In older persons, exposure to bright light and social cues is markedly diminished when compared with young adults (Ancoli-Israel, Kripke, Jones, Parker, & Hanger, 1991; Campbell, Kripke, Gillin, & Hrubovcak, 1988; Ehlers, Frank, & Kupfer, 1988). Absence of professional constraints, reduced mobility due to illness, and decreased socialization and outdoor activities are all hallmarks of old age. Therefore, diminished exposure to environmental stimuli that entrain circadian rhythms could contribute to the disruptions in circadian rhythmicity. The use of exposure to bright light in the elderly in order to reinforce circadian rhythmicity and to improve both daytime alertness and nighttime sleep has proved beneficial (Campbell, Dawson, & Anderson, 1993). Similarly, increases in physical and social activity have beneficial effects on daytime function and nighttime sleep (Naylor et al., 2000).
Sleep
Sleep Regulation
The sleep-wake cycle is one of the circadian rhythms that is driven by the circadian pacemaker and synchronized to the day-night cycle. Sleep is a rhythmic behavioral and physiologicalprocess.Historically,sleepwaslongconsideredtobea passive brain state, resulting from the withdrawal of activity in waking centers.This concept was disproven by the pioneering experiments of Bremer in 1930 and by Morruzzi and Magoun in 1940–1950, and it was recognized that sleep is actively generated by activity in specific brain regions (Rechtschaffen & Siegel, 2000). In 1953, Nathaniel Kleitman, a professor of physiology at the University of Chicago, and his graduate student Eugene Aserinsky observed the occurrence of rapid eye movements in sleeping infants. This discovery lead to the identification of two separate states within sleep: rapid eye movement (REM) sleep and non-REM (NREM) sleep.
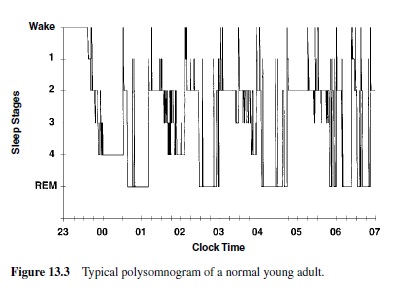
The electroencephalogram (EEG) to record brain waves, the electrooculogram (EOG) to record eye movements, and the electromyogram (EMG) to record chin muscle tension have become the primary means of monitoring the stages of human sleep. This set of recordings is called polysomnography. In 1968, Rechtschaffen and Kales defined standard criteria to score an epoch of polysomnographic recording in five stages, allowing for the determination of the macrostructure of sleep (Figure 13.3).
The EEG pattern in NREM is described as synchronous and it is conventionally subdivided into four stages. The four NREM stages can be considered as a continuum in depth of sleep, with the lowest arousal thresholds in Stage 1 and the highest in Stage 4. Often Stages 3 and 4 are lumped together and are called slow wave sleep (SWS) because the EEG is characterized by waves of high amplitude and low frequency (delta waves;0.5–4.0Hz).During SWS, the muscles are relaxed, and the autonomic system shows a parasympathetic dominance.
REM sleep is characterized by a desynchronized EEG pattern, defined by irregular waves of higher frequency and lower amplitude, similar to brain activity during waking or the lighter stage of NREM sleep. This brain pattern is associated with strong sympathetic activation, muscle atonia, and episodic bursts of rapid eye movements. The middle ear muscles are also phasically active, and both REM and middle ear muscle activity appear to be driven by phasic bursts of electrical activity that can be recorded in animals from the pons, oculomotor nuclei, thalamus, and visual cortex, and are referred to as pontine-geniculate-occipital (PGO) spikes (Rechtschaffen & Siegel, 2000).
The adult human enters sleep through NREM sleep, beginning with Stage 1, and continuing with Stages 2, 3, and 4 (Figure 13.3). Spindle activity appears in Stage 2 of NREM sleep. External stimuli can interrupt the progression from the lighter to the deeper stages of NREM sleep. About 60–80 minutes after going to sleep, the individual begins gradually to cycle back from Stage 4 through Stages 3 and 2, and then enters a period of REM. NREM and REM sleep continue to alternate through the night in cyclic fashion with a period of 90 to 110 minutes. REM sleep in the first cycle episode is usually quite short, lasting only a few minutes, but its length increases across the night; it is greatest towards the early morning hours. As the night progresses, NREM sleep becomes more shallow and REM sleep more prominent. In young adults, wakefulness after sleep onset normally accounts for less than 5% of the night, REM sleep occupies approximately 25% (aboutfour to six discrete episodes per night), while SWS constitutes approximately 20%.Thus, 50% of the night is usually spent in the lighter stages of NREM sleep.
There are specific brain mechanisms that generate NREM and REM sleep (Rechtschaffen & Siegel, 2000). Studies using lesions, electrical stimulation, and chemical stimulation have shown that the major brain regions involved in NREM sleep generation are the anterior hypothalamus and the basal forebrain. These NREM sleep mechanisms involve GABAergic inhibitory neurons that inhibit waking centers in the hypothalamus and in the midbrain. The nucleus of the solitary tract and the VLPO also participate in NREM sleep regulation (Gallopin et al., 2000; Sherin et al., 1996). It thus appears that several widely separate brain regions are involved and interact to initiate and maintain NREM sleep. During NREM sleep, slow waves in the EEG reflect the synchronized firing of thalamocortical neurons. In contrast to those involved in NREM sleep, the brain mechanisms generating REM sleep appear to be critically located in a small area of the lateral pontine tegmentum. This area contains cholinergic cells that discharge at a high rate during REM sleep (REM-on cells) and inhibit serotonergic and noradrenergic activity as well as motor neurons. Although other brain regions distant from the pons appear to participate in the control of REM sleep, the small pontine area including the REM-on cells appears to be critical. It is important to note that the three characteristics of REM sleep (i.e., desynchronized EEG, PGO waves, and atonia) can be evoked separately by cholinergic stimulation of the pons.
Humans awakened during REM report dreaming 80–90% of the times, often including elaborate visual imagery and complicated plots (Hobson, 1995; Rechtschaffen & Siegel, 2000). Awakenings from SWS are less frequently associated with oniric activity than are awakenings from REM sleep, and the dreams are generally simpler and shorter than the dreams reported after REM sleep.
In addition to the alternation of NREM and REM states, sleep is regulated by two basic processes: (a) a homeostatic process determined by the prior duration of wakefulness and largely independent of the circadian system, and (b) a circadian process that modulates sleep propensity and duration independently of homeostatic sleep pressure. The homeostatic process is thought to involve one or several putative neural sleep factors (factor S), which rise during periods of wakefulness and decay exponentially during sleep (Borbély, 1998). Sleep propensity is increased when sleep is curtailed or absent, and it is decreased in response to sleep. The timing, the amount and the intensity of SWS are primarily under the control of the homeostatic process, whereas circadian rhythmicity plays an important role in sleep timing, sleep duration, sleep consolidation, and the distribution of REM sleep.
After sleep deprivation, NREM and REM sleep are enhanced; in particular, a remarkable increase in slow waves has been documented during recovery sleep in rodents, rabbits, cats, mice, and humans (Tobler, 2000). The neuroanatomical and neurochemical basis of homeostatic sleep regulation is not entirely understood; however, it appears to involve the basal forebrain cholinergic region and adenosine, a neuromodulator whose extracellular concentrations in this region rise during sustained wakefulness and decrease during sleep (Porkka-Heiskanen et al., 1997).
It is safe to assume that all mammals sleep, even though there are large variations in the amount of sleep exhibited by different species, and sleep may be difficult to observe or recognize in some species like cetaceans (Zepelin, 2000). Sleep can usually be identified by sustained quiescence in a species-specific posture accompanied by reduced responsiveness to external stimuli. For mammals, the definition of sleep requires the additional criteria of quick reversibility to the wakeful condition (unlike hibernation or coma) and characteristic changes in the EEG activity (Zepelin, 2000). Spindling and slow waves are the hallmarks of mammalian NREM sleep, but there are considerable interspecies variations in their amplitudes. REM sleep is clearly present in all mammals studied so far, but some of the characteristics of REM sleep (frequency of eye movements and muscular atonia) as well as the relative amount of REM sleep and the duration of the REM-NREM cycle vary widely across species (Zepelin, 2000). The amount of REM sleep is positively correlated with total sleep time, and total sleep time and NREM sleep are negatively correlated with body weight—that is, heavier animals sleep less. Altricial animals (animals born in a relatively immature state and unable to care for themselves) have large amounts of REM sleep at birth; furthermore, although these amounts decrease with maturity, the amount of REM sleep in adults of altricial species are remarkably higher than in adults of precocial species (Siegel, 1995, 1999). Zepelin (2000) showed that throughout life immaturity at birth is the single best predictor of REM sleep time. It appears that REM sleep time correlates with the security of the sleep site. Predators and animals with secure sleep have greater amounts of REM sleep.
The cyclic organization of sleep is also present in birds, although the sleep cycles are much shorter than in mammalian sleep. Behavioral and EEG criteria for sleep that apply to mammals can be applied to birds. However, avian sleep has no spindling activity and lacks the differentiation in NREM sleep stages. But the main difference that distinguishes avian from mammalian sleep is the much lower percentage of REM sleep (about 5% of sleep time on average for birds, versus 15–30% in mammals) and much shorter REM periods, often less than 10 s (Zepelin, 2000). Very little is known about the sleep of fish, amphibians, and reptiles.
Ontogeny of Sleep
Sleep changes dramatically during the course of development. For the first days of life, the sleep of infants is characterized by two different types of sleep: active sleep and quiet sleep (Hobson, 1995). In the first weeks of their lives, infants spend about half of their sleeping time in active sleep. The EEG pattern of active sleep in infants is similar to that seen in adult REM sleep. However, the appearance of rapid eye movements is not associated with a complete motor paralysis as it is in adults, because the cerebral inhibitory mechanisms that block the transmission of nerve impulses to the skeletal muscles are still immature. Thus, small body movements— particularly in the fingers and toes and facial muscles—are possible.Active sleep decreases gradually during the first year, stabilizing at about 25–30% at 1 year of age. At this age body movements during REM sleep gradually disappear, and muscleatoniaisobservedasinadults.Quietsleepofinfantsisanalogous to NREM sleep in adults, but well-defined delta waves andsleepspindlescannotbediscernedduringthefirstweeksof life because electrical brain activity is still irregular and disorganized at this time. REM sleep depends on the activity of the reticular neurons in the pons, which are already relatively mature at birth; however, it is only when the cells in the cortex become extensively interconnected and when their connections to and from the thalamus are developed that the typical EEG patterns of SWS are observed and distinct sleep Stages 2, 3, and 4 develop from quiet sleep (Hobson, 1995).
The cyclic alternation of NREM-REM sleep is present from birth, but the period in the newborn is of about 50–60 minutes. A consolidated nocturnal sleep cycle is gradually acquired, and the fully developed EEG patterns of NREM sleep stages emerge over the first 2–6 months of life.Another major difference between infant and adult sleep is that in newborns the transition from wake to sleep is often accomplished through active sleep.
The declining levels of REM sleep during the first year of life are in part the result of an increase in the noradrenergic and serotonergic inhibitory control of REM and in part the result of neurons becoming less sensitive to acetylcholine, a neurotransmitter that enhances REM sleep (Hobson, 1995).
When brain structure and function achieve a level that can support the high-voltage characteristic of SWS, then Stages 3 and 4 become quite prominent in young children. In addition, their SWS is much deeper than the SWS of older adults, with a very high arousal threshold.The amount of SWS achieved per night is maximal in young children but decreases markedly with age; it decreases by nearly 40% during the second decade of life (Carskadon & Dement, 1987).
Sleep and Aging
Decreased subjective sleep quality is one of the most common health complaints of older adults (Prinz, 1995).The most consistent alterations associated with normal aging include increased number and duration of awakenings and decreased amounts of deep SWS (Benca, Obermeyer, Thisted, & Gillin, 1992; Bliwise, 1994; Feinberg, Koresko, & Heller, 1967). REM sleep appears to be relatively better preserved during aging (Benca et al., 1992; Bliwise, 1994; Ehlers & Kupfer, 1989; Feinberg, 1976; Landolt, Dijk,Achermann, & Borbely, 1996). There are gender differences in sleep quality in normal adults; premenopausal women have more SWS than do men.
The results of a recent analysis of polygraphic sleep recordings from 149 normal men ages 16–83 years are shown in Figure 13.4 (Van Cauter, Leproult, & Plat, 2000). It can be seen that age-related changes in SWS and REM sleep occur with markedly different chronologies. SWS decreases markedly from early adulthood (16–25 years) to midlife (36–50 years) and is replaced by lighter sleep (Stages 1 and 2) without significant increases in sleep fragmentation or decreases in REM sleep. The transition from midlife to late life involved an increase in wake at the expense of both NREM and REM sleep. This age-related increase in awakenings appears primarily related to an inability of maintaining NREM sleep (Dijk, Duffy, & Czeisler, 2001).The decrease in REM sleep at night has been correlated with intellectual functioning in older persons; patients with a diagnosis of probable senile dementia of Alzheimer’s type have very little REM sleep compared to healthy control subjects (Prinz et al., 1982).
Studies in laboratory rodents have also documented a decrease in SWS activity in old age (Mendelson & Bergmann, 1999). The circadian control of the sleep-wake cycle appears to be weaker in old age. A decrease in the amplitude of the circadian fluctuations of SWS and wakefulness has been observed in old rats (Rosenberg, Zepelin, & Rechtschaffen, 1979; Van Gool & Mirmiran, 1986). When older mice were compared to young mice, they spent more time asleep during the normal active period, whereas they spent more time awake during the normal sleep period (Welsh, Richardson, & Dement, 1986). Similarly, in elderly adults, nocturnal sleep becomes highly fragmented, and daytime naps become more frequent. This phenomenon is particularly prevalent in cognitively impaired older adults. Sleep disruption, nocturnal wanderings, restlessness, and daytime sleepiness constitute a major reason for institutionalization (Sanford, 1975).
Behavioral and Physiological Processes Modulated By Circadian Rhythmicity and Sleep-Wake Homeostasis
As stated earlier in this research paper, the temporal organization of behavioral and physiological variables across the 24-hour cycle reflects primarily the interaction of endogenous circadian rhythmicity and sleep-wake homeostasis. There are two features of the interaction between these two time-keeping systems that appear to be fairly unique to the human species (Van Cauter & Turek, 2001). First, human sleep is generally consolidated in a single 6- to 9-hour period, whereas fragmentation of the sleeping period is the rule in other mammals. Possibly as a result of this consolidation of the sleep period, the wake-sleep and sleep-wake transitions in humans are associated with physiological changes that are usually more marked than those observed in the animals. Second, humans are also unique in their capacity to ignore circadian signals and to maintain wakefulness for prolonged periods of time despite an increased pressure to go to sleep.
Depending on the parameter under consideration, variability across the 24-hour day may also reflect—in addition to circadian and homeostatic regulation—effects of food intake, postural changes, changes in intensity of physical activity, light-dark transitions, and other stimuli. Therefore, to facilitate the detection of circadian and homeostatic inputs, many investigators have used so-called constant routine conditions, a regimen of 24 hours or more of continuous quiet wakefulness at bed rest in dim indoor light, with constant caloric intake under the form of hourly snacks, intravenous glucose infusion at a constant rate, or constant enteral nutrition. Constant routine conditions attempt to minimize the effects of external stimuli that could alter the expression of endogenous circadian rhythmicity and sleep-wake homeostasis.
To tease apart homeostatic effects from circadian inputs, other studies have taken advantage of the fact that the circadian pacemaker takes several days to adjust to a large abrupt shift of sleep-wake and light-dark cycles. The protocols used in these shift studies allow for the effects of sleep to be observed at an abnormal circadian time and for the effects of circadian modulation to be observed in the absence of sleep. Constant routine conditions are generally applied during scheduled wakefulness periods. A third type of protocol used in studies designed to delineate the relative contributions of the circadian clock and the sleep homeostat in the temporal organization of behavioral and physiological variables are so-called forced desynchrony protocols in which the sleepwake cycles of the research subjects are scheduled to be either too long (typically 28 hours) or too short (typically 20 hours) to entrain the endogenous circadian oscillator.
Under such conditions, the circadian system free-runs with a period of approximately 24.2 hours, and the effects of sleeping and waking can be observed at all circadian phases (Dijk & Czeisler, 1995).
Alertness and Cognitive Performance
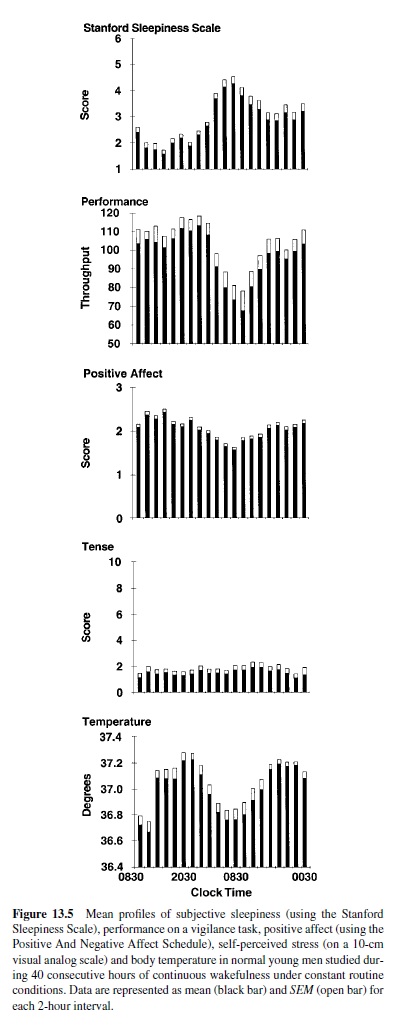
Anumber of studies under constant routine conditions have delineated the temporal variations of subjective alertness (or its antonym, subjective sleepiness) and cognitive performance (i.e., objective alertness or vigilance) on a wide variety of paper-and-pencil as well as computerized cognitive performance tasks across 24 to 40 hours of extended wakefulness (Carrier & Monk, 2000; M. P. Johnson et al., 1992; Leproult, Van Reeth, Byrne, Sturis, & Van Cauter, 1997; Monk et al., 1997; Wright, Badia, Myers, Plenzler, & Hakel, 1997). The results have been remarkably consistent and have indicated that subjective and objective alertness vary in parallel; detectable decreases start around the usual bedtime, when the circulating levels of melatonin, a hormone with sedative properties (Cajochen,Krauchi,&Wirz-Justice,1997;Sack,Hughes,Edgar,& Lewy, 1997), are increasing. The decrease in subjective and objective alertness continues until a minimum is attained in the early morning hours—that is, between 5:00 and 8:00 a.m. inthe majority of adults (Dijk, Duffy, & Czeisler, 1992; Gillberg, Kecklund, & Akerstedt, 1994; Leproult, Van Reeth, et al., 1997; Monketal., 1997). Both subjective and objective alertness then improve—despite continued wakefulness—suggesting the existence of a waking signal originating from the circadian pacemaker. The two upper panels of Figure 13.5 provide a good illustration of this typical temporal pattern in normal young men who were studied under constant routine conditions for 40 hours (Van Cauter & Turek, 2001). Subjective sleepiness was assessedhourlyusingastandardized7-pointscale,theStanford Sleepiness Scale, wherein 1 is “feeling active and vital, alert, wide awake” and 7 corresponds to “almost in reverie, sleep onset soon, lost struggle to remain awake.” Objective alertness was also estimated hourly using a computerized simple attention-dependent vigilance task. The throughput was calculated as the number of correct responses per unit of time. Maximum sleepiness coincided with minimum throughput in the early morning. Both sleepiness and performance had markedly improved by midday. Studies using forced desynchrony protocols confirmed that the temporal variations of both subjective and objective alertness during extended wakefulness are under the dual control of the homeostatic process and of a circadian rhythm promoting alertness across the usual waking period (Dijk & Edgar, 1999; Dijk & Czeisler, 1994, 1995; Dijk et al., 1992; Monk, Buysse, Reynolds, Jarrett, & Kupfer, 1992; Monk, Moline, Fookson, & Peetz, 1989).
Mood
Studies performed under constant routine conditions showed that a marked mood impairment occurs in the early morning hours when wakefulness is extended across the night (Monk et al., 1997). The lowest mood scores coincide with maximum subjective sleepiness. Similar to cognitive performance, mood then improves during the daytime hours despite continued sleep deprivation, and effects of sleep loss may no longer be detectable. The improvement in mood parallels the improvement in cognitive performance and the decrease in subjective sleepiness, as illustrated in Figure 13.5, in which the third and fourth panels from the top show the temporal variation of scores of positive affect on the Positive Affect and Negative Affect Schedule (PANAS; Clark, Watson, & Leeka, 1989) and of scores of self-perceived stress on a 10-cm visual analog scale for tense. It is apparent that the decrease in positive affect in the early morning hours following a sleepless night is not related to an increase in subjective stress.
The relative contributions of effects of time of day (i.e., circadian) as compared to effects of duration of time awake (i.e., homeostatic) were examined in healthy adults who were studied on a forced desynchrony protocol involving a 28- or 30-hour sleep-wake schedule preventing entrainment of circadian rhythmicity (Boivin et al., 1997). Mood varied primarily with circadian phase rather than with duration of prior wakefulness. However, there was a significant interaction between time of day and duration of prior wakefulness such that the impact of sleep loss on mood can be positive, nil, or negative, depending on circadian phase. In addition, the time during which the circadian variation in mood is at its nadir is prolonged with increased durations of prior wakefulness, implying that subjects who are more sleep-deprived will have greater difficulties shaking the blues of the early morning hours. These observations may be relevant to the high prevalence of mood disorders in shift workers.
Cardiovascular Function
There is a robust diurnal variation in blood pressure and heartrate across a normal 24-hour cycle when subjects are awake and active during the day and asleep at night. Figure 13.6 shows the mean 24-hour profiles of systolic and diastolic blood pressure and of heart rate measured at 10-min intervals in 31 young men who were studied in a standard social and physical environment but slept in the laboratory (Degaute, van de Borne, Linkowski, & Van Cauter, 1991). Blood pressure decreased by 10–15 mm Hg (mercury) during the sleep period, and heart rate decreased by up to 20 beats per min. Studies performed under constant routine conditions (Kerkhof, Van Dongen, & Bobbert, 1998; Van Dongen, Maislin, & Kerkhof, 2001) have revealed that there is no significant endogenous circadian variation in blood pressure. The nocturnal decline in blood pressure appears related to postural changes and sleep, with the lowest blood pressure being recorded during the deeper stages of NREM sleep. In contrast, a robust circadian variation of heart rate persists under constant routine conditions. The amplitude of the nocturnal decrease in heart rate in subjects who remain awake at bed rest across the night is approximately half of the amplitude observed when the effects of postural changes and sleep are not eliminated. So far, there is no evidence for a contribution of duration of prior wakefulness in the temporal variation of cardiovascular parameters, but few studies have rigorously addressed this issue.
Endocrine and Metabolic Regulation
A prominent feature of the endocrine system is its high degree of temporal organization. Indeed, hormonal concentrations in the peripheral circulation undergo pronounced temporal oscillations over the 24-hour cycle. The characteristics of the 24-hour patterns of hormonal secretions vary from one hormone to the other and exhibit a high degree of day-today reproducibility; thus, while one endocrine gland may be actively secreting in a complex, pulsatile pattern, another may be in a quiescent state, and this intricate temporal program of the endocrine system is likely to be of functional significance.
As for neurobehavioral and cardiovascular parameters, the 24-hour secretory profiles of many hormones and metabolic parameters are controlled by the interaction of circadian rhythmicity and sleep-wake homeostasis. The overall waveshape of the profile may also reflect—to varying degrees— modulatory effects from rhythmic and nonrhythmic factors, such as periodic food intake, postural changes, levels of physical activity, and—within the sleep state—the alternation between NREM and REM stages.
The four upper panels of Figure 13.7 illustrate mean profiles of the plasma levels of hormones secreted by the pituitary or under direct pituitary control—cortisol, thyrotropin (TSH), prolactin (PRL), and growth hormone (GH)—observed in normal subjects who were studied before and during an abrupt 12-hour shift of the sleep-wake and dark-light cycle. The study period extended over a 53-hour span and included an 8-hour period of nocturnal sleep starting at 11:00 p.m., a 28-hour period of continuous wakefulness, and a daytime period of recovery sleep starting 12 hours out of phase with the usual bedtime—that is, at 11:00 a.m. (Van Cauter et al., 1991). Constant routine conditions were enforced during scheduled waking periods. To allow for the observation of circadian variations in glucose tolerance in the absence of the effects of feeding and fasting, caloric intake was exclusively in the form of an intravenous glucose infusion at a constant rate. The two lower panels of Figure 13.7 illustrate the temporal profiles of blood glucose and insulin secretion rates (ISR).
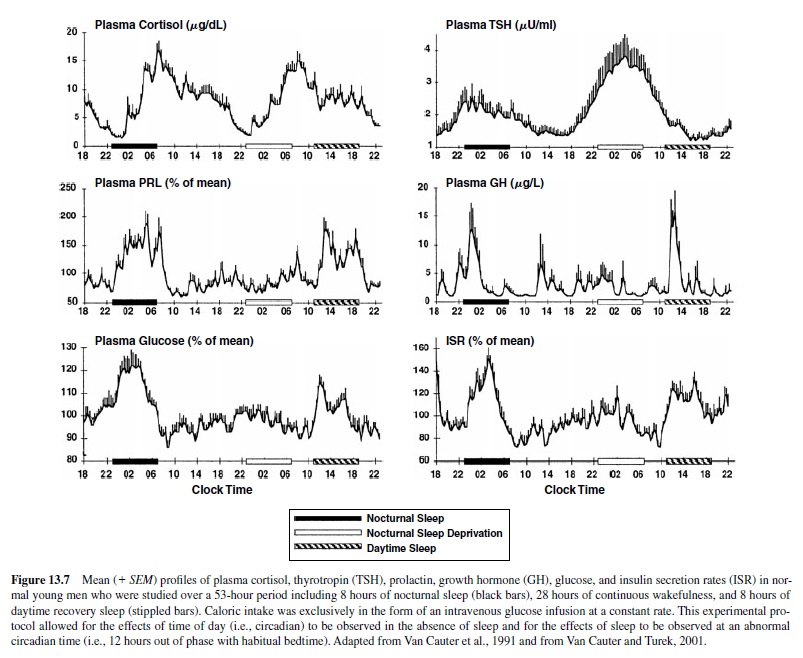
The upper panel of Figure 13.7 show that the 12-hour shift of the sleep-wake schedule had relatively little effect on the waveshape of the cortisol profile—consistent with numerous studies indicating that the temporal organization of the hypothalamic-pituitary axis is primarily dependent on circadian timing. Nevertheless, modest modulatory effects of wake-sleep and sleep-wake transitions on corticotropic activity have been clearly demonstrated. Sleep onset is reliably associated with a short-term inhibition of cortisol secretion (Born, Muth, & Fehm, 1988; Van Cauter et al., 1991; Weitzman, Zimmerman, Czeisler, & Ronda, 1983), although this effect may not be detectable when sleep is initiated at the time of the daily maximum of corticotropic activity—that is, in the morning (Weibel, Follenius, Spiegel, Ehrhart, & Brandenberger, 1995). Under conditions of normal nocturnal bedtimes, because cortisol secretion is already quiescent in the late evening, this inhibitory effect simply prolongs the quiescent period of cortisol secretion. This inhibition of hypothalamic-pituitary-adrenal (HPA) activity during sleep occurs during SWS (Follenius, Brandenberger, Bardasept, Libert, & Ehrhart, 1992). Conversely, awakening at the end of the sleep period is consistently followed by a pulse of cortisol secretion. Several studies have shown that awakenings interrupting the sleep period consistently trigger pulses of cortisol secretion (Van Cauter, Copinschi, & Turek, 2001). Thus, sleep fragmentation—as it occurs in normal aging and in persons with insomnia—is associated with increased nocturnal corticotropic activity (Vgontzas et al., 2001).
The contribution of both circadian inputs and sleepdependent modulation can easily be recognized in the temporal profiles of TSH concentrations. Low and relatively stable daytime TSH levels are followed by a rapid elevation starting in the early evening and peaking around the time of habitual sleep onset (second panel from the top in Figure 13.7). The timing of the nocturnal TSH elevation appears to accurately reflect circadian timing and shifts with the rhythms of body temperature and melatonin (Allan & Czeisler, 1994; Van Cauter et al., 1994). The normal sleep period is associated with a progressive decline in TSH levels, which is related to a consistent decrease of TSH levels during SWS. Daytime basal values resume shortly after morning awakening. The inhibitory effect of sleep on TSH secretion becomes clearly apparent when nocturnal levels are observed during acute sleep deprivation—then TSH secretion is increased by as much as 200% over the levels observed during nocturnal sleep. When sleep occurs during daytime hours, TSH secretion is not suppressed significantly below normal daytime levels.
Contrasting with the strong circadian modulation of activity in the corticotropic and thyrotropic axes, the temporal organization of the release of prolactin and GH is primarily driven by sleep-wake homeostasis (Van Cauter, Plat, & Copinschi, 1998). As can be seen from Figure 13.7, a large portion of the daily output of both hormones occurs during sleep, and the secretory pattern shifts immediately following the shift of the sleep-wake cycle. In both men and women, prolactin levels during sleep correspond to an average increase of more than 200% above daytime levels. Increases in prolactin secretion during sleep are consistently associated with SWS. A modest circadian component in the temporal organization of prolactin secretion can sometimes be detected—particularly in women (Waldstreicher et al., 1996)—but sleep onset, regardless of time of day, is invariably associated with prolactin release. Decreased dopaminergic inhibition of prolactin is the most likely mechanism underlying the sleep-related prolactin elevation. Nocturnal prolactin release is decreased in older adults, consistent with the decrease in sleep intensity and consolidation.
For GH, there is a consistent temporal association between increased pulsatile release and periods of SWS, particularly during the first NREM-REM cycle (Van Cauter et al., 1998). In men, 60–70% of the daily GH secretion occurs during the first hours of sleep. In women, daytime GH pulses are more frequent. Sleep onset will elicit a pulse in GH secretion whether sleep is advanced, delayed, or interrupted and reinitiated. Whereas sleep-wake homeostasis is clearly the major factor controlling the temporal organization of GH release, the inhibitory control of GH secretion by somatostatin appears to be influenced by circadian rhythmicity, with lower nocturnal somatostatinergic tone facilitating growth hormone releasing hormone (GHRH)-dependent release of GH during sleep. Sleep fragmentation and reduced amounts of SWS will generally decrease nocturnal GH secretion. In healthy older adults, the total amount of GH secreted over the 24-hour span is often less than one third of the daily output of young men; the amount of SWS is similarly reduced (Van Cauter et al., 2000; van Coevorden et al., 1991). There is a remarkable parallelism between chronological alterations of GH levels and SWS across adulthood, and correlative evidence suggests that age-related alterations in the somatotropic axis are partly caused by reduced sleep quality.
As seen in the two lower panels of Figure13.7—despite the fact that glucose was infused at a constant rate throughout the study period—there were robust changes in plasma glucose levels and insulin secretion rates. Under these experimental conditions, hepatic glucose production is markedly suppressed; therefore, changes in plasma glucose levels reflect primarily changes in glucose utilization (Van Cauter, Polonsky, & Scheen, 1997). Glucose levels increase from morning to evening, indicating a decrease in glucose tolerance as the day progresses, reach a maximum around midsleep, and then decrease toward morning values. The pattern of insulin secretion parallels the changes in glucose levels. The nocturnal glucose and insulin elevations readily shift with the shift of sleep-wake schedule, indicating a major effect of the sleep state regardless of time of day (Van Cauter et al., 1991). Nevertheless, in the absence of sleep, an elevation of glucose and insulin secretion from morning until the early part of the usual sleep period can be clearly seen (Figure 13.7). Detailed studies have indicated that when sleep occurs at the normal circadian time, decreased glucose tolerance during the first part of the night is due to decreased glucose utilization in both peripheral tissues—a result of muscle relaxation, antiinsulin-like effects of sleep-onset GH secretion, and reduced cerebral glucose uptake during SWS (Van Cauter et al., 1997). During the second part of the night, these effects subside as sleep becomes shallow, movements and awakenings increase, REM sleep is predominant, and GH secretion is minimal. Thus, complex interactions of circadian and sleep effects— partly mediated by cortisol and GH—result in a consistent pattern of changes of set-point of glucose regulation over the 24-hour period.
Conditions Ofabnormal Sleepand Circadian Regulation
Behavioral Alterations
Acute Sleep Deprivation and Chronic Sleep Curtailment
Immediate effects of acute total sleep deprivation on a variety of neurobehavioral and physiological parameters are well recognized. The adverse effects on subjective alertness, cognitive performance, and mood have been described in previous sections of this research paper. In the absence of sleep, the nocturnal declines in body temperature and heart rate are considerably dampened, and blood pressure does not dip. As illustrated in Figure 13.7, the secretions of GH and prolactin normally associated with sleep onset are largely suppressed.
Until recently, the only well-documented effects of sleep loss on human physiological function were the absence of immediate responses to sleep-wake transitions in conditions of total sleep deprivation and changes in markers of immune function that become evident after prolonged sleep deprivation (Dinges, Douglas, Hamarman, Zaugg, & Kapoor, 1995). Because recovery sleep on the subsequent night is associated with normal temperature and cardiovascular changes and because rebound secretions of hormones normally released during sleep are usually observed, it has often been implicitly assumed that there is no net effect of sleep loss on human physiological function and endocrine status. Nevertheless, acute sleep deprivation—whether total or partial—is associated with an alteration in hypothalamic-pituitary-adrenal (HPA) function on the following day, consisting of an elevation of evening cortisol concentrations (Leproult, Copinschi, Buxton, & Van Cauter, 1997). Because deleterious central as well as metabolic effects of HPA hyperactivity are more pronounced at the time of the usual trough of the rhythm (i.e., in the evening) than at the time of the peak (i.e., in the morning), modest elevations in evening cortisol levels could facilitate the development of central as well as peripheral disturbances associated with glucocorticoid excess, such as memory deficits and insulin resistance (Dallman et al., 1993; McEwen & Sapolsky, 1995). Morning oral glucose tolerance is decreased after a sleepless night as compared to a normal night (VanHelder, Symons, & Radomski, 1993).
Although there have been numerous studies of the effects of acute total sleep deprivation, the much more common reallife condition of chronic partial sleep curtailment has received much less attention. Voluntary sleep restriction is, however, an increasingly common behavior in industrialized societies. Due to socioeconomic and cultural pressures, many individuals tend to curtail sleep to the shortest amount tolerable (Bliwise, 1996; Bonnet & Arand, 1995; Wehr et al., 1993). Sleep duration in America decreased from 9.1 hours in 1910 to less than 7 hours in 2000. In support of sleep curtailment as both harmless and efficient, it has been proposed that a socalled normal (approximately 8-hour) night of sleep is composed of core sleep, a 4- to 5-hour period including most of deep NREM sleep, and optional sleep, which could—with adequate amounts of motivation—be progressively removed without producing increased daytime sleepiness, mood changes, or detectable decrements in cognitive function (Horne, 1988). More recent studies have disproved this concept and demonstrated that 7–8 consecutive days of sleep curtailment by as little as 2–3 hours per night results in marked decrements in mood and performance (Dinges et al., 1997). Although the consequences of endemic sleep loss for human performance and safety have recently received public attention, it is generally thought that physiological function and health are affected little or not at all.
A recent study from our laboratory examined metabolic and hormonal parameters in healthy young adults studied after 1 week of bedtimes restricted to 4 hours per night and then after 1 week of bedtimes extended to 12 hours per night (Spiegel, Leproult, & Van Cauter, 1999). The upper panels of Figure 13.8 compare the glucose and insulin responses to an intravenous glucose tolerance test performed at 9:00 a.m. on the 5th day of sleep restriction and the 5th day of sleep extension. The parameters of glucose tolerance in the state of sleep debt were consistent with the clinical condition of impaired glucose tolerance. Insulin secretion during the first phase of the response was markedly dampened, as occurs in the early stages of diabetes. The lower panels of Figure 13.8 compares the profiles of sympatho-vagal balance, evening cortisol levels, and nocturnal GH secretion for the two bedtime conditions. In the state of sleep debt (compared to the fully rested condition), sympathetic nervous activity was elevated, evening cortisol levels were higher, and nocturnal GH secretion was prolonged (Spiegel et al., 2000). All three alterations are likely to have contributed to the decrease in glucose tolerance. Decreased brain glucose utilization, which has been documented in sleep-deprived subjects by PET (positron-emission tomography) studies (Thomas et al., 2000), probably contributed also to this metabolic alteration. These results indicate that partial sleep loss under chronic conditions could have long-term adverse health effects and accelerate the development or increase the severity of agerelated diseases such as diabetes and hypertension.
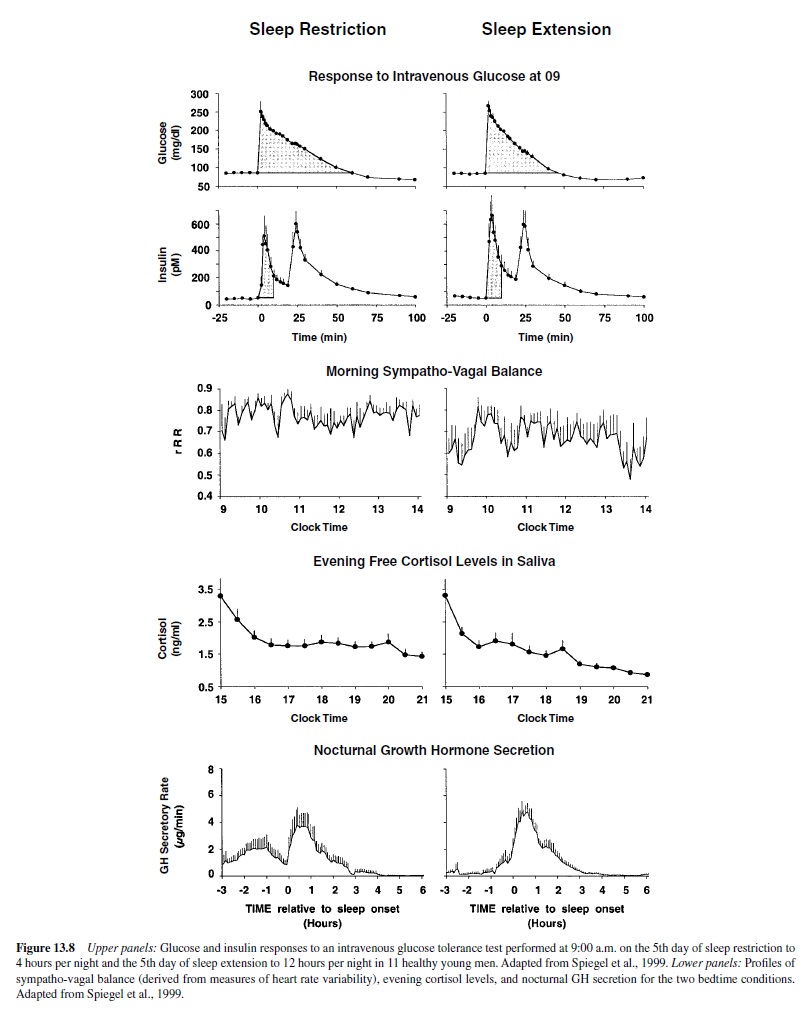
Profound alterations of neurobehavioral function were also observed after 1 week of sleep restriction. Ratings of subjective sleepiness and reaction times on tests of cognitive performance were markedly increased, whereas mood scores were significantly lower. These alterations were more severe in the morning than they were later in the day, and a robust diurnal variation of mood and vigilance similar to that seen in depressed subjects was apparent. Remarkably, many of the endocrine, neurobehavioral, and metabolic alterations observed in these healthy young adults after 1 week of severe sleep curtailment were similar to well-documented markers of aging.
Shift Work
Although shift work has been part of modern life for several decades and is voluntarily accepted by millions of workers, it is a major health hazard involving an increased risk of cardiovascular illness, gastrointestinal disorders, insomnia, mood disorders, and infertility (Knutsson, Akerstedt, Jonsson, & Orth-Gomer, 1986). The consequences of shift work are associated with the chronic misalignment of physiological circadian rhythms, the activity-rest cycle, and the unavoidable sleep loss that results. Shift work creates conditions in which some zeitgebers (e.g., an artificial LD cycle) and additional phase-setting factors such as the rest-activity cycle, are shifted while others remain unaltered—for example, the natural LD cycle and the routines of family and social life. Nearly all shift work schedules involve days off every few days, and the worker then usually attempts to revert to daytime activity and nighttime sleep. Thus, shift workers live in an unstable situation of conflicting zeitgebers that almost never allow a complete alignment of endogenous circadian rhythmicity with the sleep-wake/rest-activity cycle and is characterized by a chronic internal desynchrony (Weibel, Spiegel, Follenius, Ehrhart, & Brandenberger, 1996). Components of overt rhythms that are controlled primarily by sleep-wake homeostasis (such as sleep-related GH and prolactin secretion) adapt to the shifted schedule, whereas components reflecting circadian timing (such as the diurnal variation of cortisol secretion) showed little (if any) adaptation. Recent polls have indicated that shift workers are also among the most sleep-deprived segment of the population, with mean sleep duration averaging only 6.5 hours in 2000 (E. O. Johnson, 2000). Thus, in addition to circadian misalignment, shift work is also a condition of chronic sleep debt. Not surprisingly, night work is associated with deficits in performance and vigilance, diminished productivity, and increased accident rates.
The need for strategies to cope with shift work is obvious, particularly in view of the fact that the chronic use of hypnotics in shift work has deleterious rather than beneficial effects. Scheduled exposure to bright light during night work and complete darkness during daytime sleep can accelerate circadian adjustment to a night schedule and improve nighttime alertness and performance (Czeisler et al., 1990; Dawson & Campbell, 1991; Eastman, Stewart, Mahoney, Liu, & Fogg, 1994). This strategy is of limited use for the majority of shift workers, who rarely stay long enough on a given schedule to benefit from this type of intervention.Another experimental approach for improving adaptation to night work is the use of melatonin (Folkard,Arendt, & Clark, 1993). This treatment theoretically combines the phase-shifting effects of the drug with the facilitation of daytime sleep. However, the few controlled trials performed have given mixed results; moreover, optimization of timing of administration, dosage, and formulation need to be evaluated further (Arendt, Skene, Middleton, Lockley, & Deacon, 1997).
Transmeridian Flights and Jet Lag
Upon arrival of a transmeridian flight, the travelers are confronted with a desynchronization between their internal circadian rhythms and the periodicity of the new external environment. The lack of synchronization is associated with symptoms of fatigue, impaired mental and psychomotor performance, subjective discomfort, sleep disturbances, and gastrointestinal disorders. Phase-shifts of human circadian rhythms have been studied after actual jet lag as well as after abrupt displacements of the LD cycle, the sleep-wake cycle, or both, designed to reproduce in the laboratory the desynchrony that occurs in jet lag.After an abrupt shift caused by real or simulated jet lag, adaptation is not immediate and requires several days. The rate at which reentrainment occurs differs among variables, and in general it is slower for overt rhythms that are strongly dependent on the circadian system (such as those of cortisol and melatonin secretions) than it is for those that are markedly modulated by sleep-wake homeostasis (such as prolactin and GH secretions).
As a result of differences in the rate of adaptation for different physiological subsystems, abnormal phase relationships between overt rhythms occur during the period of adaptation. Therefore, jet lag involves not only desynchronization between internal and external rhythms, but also a perturbation of internal temporal organization of physiological functions. The overall rate of adaptation depends on the strength of the zeitgebers, and it can be as low as half an hour per day or as high as 3 hours per day. Moreover, the rate of adaptation is not constant: Adaptation occurs at a faster pace during the first few days and progresses at a slower rate thereafter (Aschoff, Hoffmann, Pohl, & Wever, 1975). The rate of adaptation is also dependent on the direction of the shift: Adaptation usually occurs faster after a delay shift (traveling westward) than after an advance shift (traveling eastward; Aschoff et al., 1975). This difference is generally believed to be due to the fact that the endogenous circadian period of humans is longer than 24 hours; therefore, adjustment by delays ismoreeasilyachievedthanisadjustmentbyadvances.There is strong evidence to suggest that reentrainment after a transmeridian flight is facilitated by exposure to bright light at appropriate circadian phases (Hirschfeld et al., 1996; Wever, 1986). The major difficulty is the timing of light exposure on successive days after the shift. As circadian phase dynamically changes, it is practically impossible (given our current state of knowledge) to predict the optimal timings of light exposurebeyondthefirstdayaftertheshift.Itiswidelybelieved that adherence to the local social and meal schedule upon arrival will accelerate the adaptation to jet lag, but this has not been rigorously demonstrated. Laboratory studies suggest that physical exercise scheduled during the period corresponding to the nighttime prior to travel is capable of phasedelaying human rhythms and therefore facilitates adaptation to a delay (i.e., westward) shift (Buxton et al., 1997; Van Reeth et al., 1994). Appropriately timed use of benzodiazepine hypnotics can also facilitate adaptation to jet lag (Buxton, Copinschi, Van Onderbergen, Karrison, & Van Cauter, 2000).
Pathological Alterations
Insomnia
Insomnia is the experience or complaint of poor quality or quantity of sleep. Three categories of insomnia can be distinguished: onset insomnia (difficulty falling asleep), maintenance insomnia (waking up frequently during the night), and termination insomnia (waking up too early and not being able to fall asleep again). Insomnia is one of the most common complaints encountered by physicians working in primary care settings. In Western industrial countries, approximately one third of the adult population reports at least occasional difficulties sleeping (Ancoli-Israel & Roth, 1999). The prevalence of chronic insomnia is approximately 10% (Hajak, 2000; E. O. Johnson, 2000; Stepanski, Zorick, Roehrs, & Roth, 2000) and increases with age: Approximately one third of subjects older than 65 years are estimated to suffer from chronic insomnia. Insomnia occurs about 1.5 times more often in women than in men; this is especially true for perimenopausal and postmenopausal women (Hublin, Kaprio, Partinen, & Koskenvuo, 2001).
Untreated insomnia may have severe consequences for the health and well-being of the patient. Insomnia is frequently associated with sleepiness, fatigue, impairments in daytime functioning, inability to concentrate, lack of alertness, memory troubles, and mood disorders (DSM-IV, 1994). Individuals with chronic insomnia report elevated levels of stress, depression, anxiety, muscle aches, and medical illnesses (Aldrich, 2000). In addition, those with chronic insomnia reported 2.5 times more fatigue-related car accidents than did good sleepers. A recent study has demonstrated that the HPA axis is hyperactive in persons with insomnia (Vgontzas et al., 2001). As in studies of experimental sleep deprivation or sleep restriction, cortisol levels are higher in the evening and during the night. This endocrine alteration could play a role in the mood and memory disturbances that are frequently associated with insomnia, as well as promote the development of insulin resistance.
Obstructive Sleep Apnea
Obstructive sleep apnea syndrome (OSAS) is characterized by episodes of complete or partial pharyngeal obstruction during sleep accompanied by excessive daytime sleepiness. The main symptoms of OSAS are loud and irregular snoring, restless and nonrefreshing sleep, and daytime fatigue. Acharacteristic pattern of apneic episodes is brief gasps or loud snoring that alternate with episodes of silence typically lasting 20–30 s. Breathing is impaired because there is a reduction in muscle tone at sleep onset and a change in the central control of respiration during sleep (Roth & Roehrs, 2000). These two events result in an abnormality in the anatomy of the upper airway—specifically, the narrowing of the pharynx. The prevalence of the syndrome is between 2% and 5% of the population, but it may be as much as 8% in men 40–59 years of age (Hublin et al., 2001). The strongest risk factors for OSAS are obesity and age of more than 65 years; male gender has also been shown to be a risk factor. Sleep apnea is a major risk factor for hypertension, heart attacks, and stroke. The severe daytime sleepiness that is often associated with OSAS results in a major safety risk for the patient and his or her environment.
The established treatment for moderate to severe OSAS is continuous positive airway pressure (CPAP) therapy via nasal masks. OSAS causes sleep fragmentation and is usually associated with suppression of SWS sleep, REM sleep, or both. Successful treatment of OSAS (as with CPAP) produces a more consolidated sleep pattern with huge rebounds of SWS or REM sleep and prompt relief of daytime sleepiness (Carskadon & Dement, 2000). In addition, treatment with CPAP resulted in a clear increase in the amount of GH secreted during the first few hours of sleep (Cooper, White, Ashworth, Alberti, & Gibson, 1995; Saini et al., 1993).
Delayed and Advanced Sleep Phase Syndrome
Delayed sleep phase syndrome is considered a circadian rhythm sleep disorder. It is characterized by a chronic inability to fall asleep at a normal bedtime and to awake in the morning (Weitzman et al., 1981). It has been proposed that the basis of this disorder is an inability to advance the timing of the circadian sleep-wake cycle after it has been delayed to an abnormal phase by late bedtimes during a weekend or after transmeridian travel. Nonpharmacological chronotherapy involving repeated scheduled exposure to bright light is the treatment of choice for this disorder (Rosenthal et al., 1990).
In contrast, in the advanced sleep phase syndrome, the timing of the major sleep episode is advanced in relation to normal bedtime, resulting in symptoms of extreme evening sleepiness and early morning awakening. The complaint of waking up earlier than desired is common in older adults, but extreme cases of advance sleep phase are very rare. Two recent studies (Jones et al., 1999; Reid et al., 2001) have described families with the advanced sleep phase syndrome; both have concluded that the trait segregates with an autosomal dominant mode. At least one form of familial advanced sleep phase syndrome appears related to an abnormally short endogenous circadian period (Jones et al., 1999) apparently caused by a mutation altering the function of a circadian clock gene, hPer2 (Toh et al., 2001).
Conclusions
The present review has attempted to summarize the evidence indicating that every facet of our neurobiology and psychology undergoes consistent temporal variations on a 24-hour time scale. These temporal variations are largely determined by the interaction of two central nervous systems that keep track of time—circadian rhythmicity and sleep homeostasis. The impact of these two time-keeping systems is ubiquitous across all aspects of human function and—for some systems—the combined effects of circadian rhythmicity and sleep regulation may result in modulatory effects that are as large as those associated with the transition from normal to pathological function. The advent of artificial light and the emergence of a 24-hour society in industrialized countries have resulted in major disruptions of the natural control of the temporal organization of human activities, with major consequences for mental and physical well-being, safety, and productivity. During recent years, there has been a rapid increase in the understanding of the neuroanatomical and molecular mechanisms underlying the generation and transmission of the circadian signal and the initiation, duration, and quality of sleep. These advances offer the hope that strategies that improve sleep quality—particularly in older adults and in those suffering from chronic sleep loss—will have benefits for mental and physical health. The rapid progress in basic circadian biology holds the promise that conditions of circadian misalignment such as those that occur in jet lag and shift work will become treatable in a not-so-distant future.
Bibliography:
- Aldrich, M. (2000). Cardinal manifestations of sleep disorders. In M. Kryger, T. Roth, & W. Dement (Eds.), Principles and practices of sleep medicine (3rd ed., pp. 526–534). Philadelphia: W. B. Saunders.
- Allan, J. S., & Czeisler, C. A. (1994). Persistence of the circadian thyrotropin rhythm under constant conditions and after lightinduced shifts of circadian phase. Journal of Clinical Endocrinology and Metabolism, 79, 508–512.
- Ancoli-Israel, S., Kripke, D. F., Jones, D. W., Parker, L., & Hanger, M. A. (1991). 24-hour sleep and light rhythms in nursing home patients. Sleep Research, 20A,
- Ancoli-Israel, S., & Roth, T. (1999). Characteristics of insomnia in the United States: I. Results of the 1991 National Sleep Foundation Survey. Sleep, 22(Suppl. 2), S347–S353.
- Arendt, J., Skene, D. J., Middleton, B., Lockley, S. W., & Deacon, S. (1997). Efficacy of melatonin treatment in jet lag, shift work, and blindness. Journal of Biological Rhythms, 12(6), 604–617.
- Aschoff, J., Hoffmann, K., Pohl, H., & Wever, R. (1975). Reentrainment of circadian rhythms after phase-shifts of the Zeitgeber. Chronobiologia, 2(1), 23–78.
- Aston-Jones, G., Chen, S., Zhu, Y., & Oshinsky, M. L. (2001). A neural circuit for circadian regulation of arousal. Nature Neuroscience, 4(7), 732–738.
- Benca, R. M., Obermeyer, W. H., Thisted, R. A., & Gillin, J. C. (1992). Sleep and psychiatric disorders. Archives of General Psychiatry, 49, 651–668.
- Bliwise, D. L. (1994). Normal aging. In M. H. Kryger, T. Roth, & W. C. Dement (Eds.), Principles and practices of sleep medicine (pp. 26–39). Philadelphia: W. B. Saunders.
- Bliwise, D. L. (1996). Historical change in the report of daytime fatigue. Sleep, 19, 462–464.
- Boivin, D. B., Czeisler, C. A., Dijk, D. J., Duffy, J. F., Folkard, S., Minors, D. S., Totterdell, P., & Waterhouse, J. M. (1997). Complex interaction of the sleep-wake cycle and circadian phase modulates mood in healthy subjects. Archives of General Psychiatry, 54, 145–152.
- Bonnet, M., & Arand, D. (1995). We are chronically sleep deprived. Sleep, 18, 908–911.
- Borbély, A. A. (1998). Processes underlying sleep regulation. Hormone Research, 49, 114–117.
- Born, J., Muth, S., & Fehm, H. L. (1988). The significance of sleep onset and slow wave sleep for nocturnal release of growth hormone (GH) and cortisol. Psychoneuroendocrinology, 13, 233–243.
- Brock, M. A. (1991). Chronobiology and aging. Journal of American Geriatric Society, 39, 74–91.
- Buxton, O. M., Copinschi, G., Van Onderbergen, A., Karrison, T. G., & Van Cauter, E. (2000). A benzodiazepine hypnotic facilitates adaptation of circadian rhythms and sleep-wake homeostasis to an eight hour delay shift simulating westward jet lag. Sleep, 23(7), 915–927.
- Buxton, O. M., Frank, S. A., L’Hermite-Balériaux, M., Leproult, R., Turek, F. W., & Van Cauter, E. (1997). Roles of intensity and duration of nocturnal exercise in causing phase-shifts of human circadian rhythms. American Journal of Physiology (Endocrinology and Metabolism), 273, E536–E542.
- Cajochen, C., Krauchi, K., & Wirz-Justice, A. (1997). The acute soporific action of daytime melatonin administration: Effects on the EEG during wakefulness and subjective alertness. Journal of Biological Rhythms, 12, 636–643.
- Campbell, S. S., Dawson, D., &Anderson, M.W. (1993).Alleviation of sleep maintenance insomnia with timed exposure to bright light. Journal of American Geriatric Society, 41, 829–836.
- Campbell, S. S., Kripke, D. F., Gillin, J. C., & Hrubovcak, J. C. (1988). Exposure to light in healthy elderly subjects and Alzheimer’s patients. Physiology and Behavior, 42, 141–144.
- Carrier,J.,&Monk,T.H.(2000).Circadianrhythmsofperformance: New trends. Chronobiology International, 17, 719–732.
- Carrier, J., Monk, T. H., Buysse, D. J., & Kupfer, D. J. (1996). Amplitude reduction of the circadian temperature and sleep rhythms in the elderly. Chronobiology International, 13(5), 373–386.
- Carskadon, M. A., & Dement, W. C. (1987). Sleepiness in the normal adolescent. In C. Guilleminault (Ed.), Sleep and its disorders in children (pp. 53–66). New York: Raven Press.
- Carskadon, M., & Dement, W. C. (2000). Normal human sleep: An overview. In M. Kryger, T. Roth, & W. C. Dement (Eds.), Principles and practices of sleep medicine (3rd ed., pp. 15–25). Philadelphia: W. B. Saunders.
- Clark, L. A., Watson, D., & Leeka, J. (1989). Diurnal variation in the positive affects. Motivation and Emotion, 13, 205–234.
- Cooper, B. G., White, J. E., Ashworth, L. A., Alberti, K. G., & Gibson, G. J. (1995). Hormonal and metabolic profiles in subjects with obstructive sleep apnea syndrome and the acute effects of nasal continuous positive airway pressure (CPAP) treatment. Sleep, 18(3), 172–179.
- Copinschi, G., Leproult, R., & Van Cauter, E. (2001). Sleep and hormonal rhythms in humans. In P. R. Hof & C. V. Mobbs (Eds.), Functional neurobiology of aging (pp. 855–868). San Diego, CA: Academic Press.
- Czeisler, C. A., Chiasera, A. J., & Duffy, J. F. (1991). Research on sleep, circadian rhythms and aging: Applications to manned spaceflight. Experimental Gerontology, 26, 217–232.
- Czeisler, C. A., Duffy, J. F., Shanahan, T. L., Brown, E. N., Mitchell, J. F., Rimmer, D. W., Ronda, J. M., Silva, E. J., Allan, J. S., Emens, J. S., Dijk, D.-J., & Kronauer, R. E. (1999). Stability, precision, and near-24-hour period of the human circadian pacemaker. Science, 284, 2177–2181.
- Czeisler, C. A., Johnson, M. P., Duffy, J. F., Brown, E. N., Ronda, J. M., & Kronauer, R. E. (1990). Exposure to bright light and darkness to treat physiologic maladaptation to night work. New England Journal of Medicine, 322(18), 1253–1259.
- Dallman, M. F., Strack, A. L., Akana, S. F., Bradbury, M. J., Hanson, E. S., Scribner, K. A., & Smith, M. (1993). Feast and famine: Critical role of glucocorticoids with insulin in daily energy flow. Frontiers in Neuroendocrinology, 14, 303–347.
- Davis, F. C., Frank, M. G., & Heller, C. H. (1999). Ontogeny of sleep and circadian rhythms. In F. W. Turek & P. C. Zee (Eds.), Regulation of sleep and circadian rhythms (pp. 19–62). New York: Marcel Dekker.
- Dawson, D., & Campbell, S. S. (1991). Timed exposure to bright light improves sleep and alertness during simulated night shifts. Sleep, 14, 511–516.
- Degaute, J. P., van de Borne, P., Linkowski, P., & Van Cauter, E. (1991). Quantitative analysis of the 24-hour blood pressure and heart rate patterns in young men. Hypertension, 18, 199–210.
- Dijk, D.-J., & Czeisler, C. A. (1994). Paradoxical timing of the circadian rhythm of sleep propensity serves to consolidate sleep and wakefulness in humans. Neuroscience Letters, 166, 63–68.
- Dijk, D.-J., & Czeisler, C. A. (1995). Contribution of the circadian pacemaker and the sleep homeostat to sleep propensity, sleep structure, electroencephalographic slow waves, and sleep spindle activity in humans. Journal of Neuroscience, 15, 3526–3538.
- Dijk, D.-J., Duffy, J. F., & Czeisler, C. A. (1992). Circadian and sleep/wake dependent aspects of subjective alertness and cognitive performance. Journal of Sleep Research, 1, 112–117.
- Dijk, D.-J., Duffy, J. F., & Czeisler, C. A. (2001). Age-related increase in awakenings: impaired consolidation of nonREM sleep at all circadian phases. Sleep, 24(5), 565–577.
- Dijk, D.-J., & Edgar, D. M. (1999). Circadian and homeostatic control of wakefulness and sleep. In P. C. Zee & F. W. Turek (Eds.), Regulation of sleep and circadian rhythms (pp. 111–147). New York: Marcel Dekker.
- Dinges, D. F., Douglas, S. D., Hamarman, S., Zaugg, L., & Kapoor, S. (1995). Sleep deprivation and human immune function. Advances in Neuroimmunology, 5, 97–110.
- Dinges, D. F., Pack, F., Williams, K., Gillen, K., Powell, J., Ott, G., Aptowicz, C., & Pack, A. (1997). Cumulative sleepiness, mood disturbance, and psychomotor vigilance performance decrements during a week of sleep restricted to 4–5 hours per night. Sleep, 20, 267–277.
- Dunlap, J. C. (2000). A new slice on an old problem. Nature Neuroscience, 3(4), 305–306.
- Eastman, C. I., Stewart, K. T., Mahoney, M. P., Liu, L., & Fogg, L. F. (1994). Dark goggles and bright light improve circadian rhythm adaptation to night-shift work. Sleep, 17(6), 535–543.
- Ehlers, C. L., Frank, E., & Kupfer, D. J. (1988). Social zeitgebers and biological rhythms. Archives of General Psychiatry, 45, 948–952.
- Ehlers, C. L., & Kupfer, D. J. (1989). Effects of age on delta and REM sleep parameters. Electroencephalography and Clinical Neurophysiology, 72, 118–125.
- Feinberg, I. (1976). Functional implications of changes in sleep physiology with age. In R. D. Terry & S. Gershon (Eds.), Neurobiology of aging (pp. 23–41). New York: Raven Press.
- Feinberg, I., Koresko, R. L., & Heller, N. (1967). EEG sleep patterns as a function of normal and pathological aging in man. Psychiatry Research, 5, 107–144.
- Folkard, S., Arendt, J., & Clark, M. (1993). Can melatonin improve shift workers’ tolerance of the night shift? Some preliminary findings. Chronobiology International, 10, 315–320.
- Follenius, M., Brandenberger, G., Bardasept, J., Libert, J., & Ehrhart, J. (1992). Nocturnal cortisol release in relation to sleep structure. Sleep, 15, 21–27.
- Gallopin, T., Fort, P., Eggermann, E., Cauli, B., Luppi, P. H., Rossier, J., Audinat, E., Mühlethaler, M., & Serafin, M. (2000). Identification of sleep-promoting neurons in vitro. Nature, 404, 992–995.
- Gillberg, M., Kecklund, G., & Akerstedt, T. (1994). Relations between performance and subjective ratings of sleepiness during a night awake. Sleep, 17, 236–241.
- Hajak, G. (2000). Insomnia in primary care. Sleep, 23(Suppl. 3), S54–S63.
- Hirschfeld, U., Moreno-Reyes, R., Akseki, E., L’Hermite-Balériaux, M., Leproult, R., Copinschi, G., & Van Cauter, E. (1996). Progressive elevation of plasma thyrotropin during adaptation to simulated jet lag: Effects of treatment with bright light or zolpidem. Journal of Clinical Endocrinology and Metabolism, 81, 3270–3277.
- Hobson, J. A. (1995). New York: Scientific American Library.
- Horne, J. (1988). Why we sleep. Oxford, UK: Oxford University Press.
- Hublin, C., Kaprio, J., Partinen, M., & Koskenvuo, M. (2001). Insufficient sleep: a population-based study in adults. Sleep, 24(4), 392–400.
- Johnson, E. O. (2000). Sleep in America: 2000. Washington, DC: National Sleep Foundation.
- Johnson, M. P., Duffy, J. F., Dijk, D. J., Ronda, J. M., Dyal, C. M., & Czeisler, C. A. (1992). Short-term memory, alertness and performance: A reappraisal of their relationship to body temperature. Journal of Sleep Research, 1, 24–29.
- Jones, C. R., Campbell, S. S., Zone, S. E., Cooper, F., DeSano, A., Murphy, P. J., Jones, B., Czajkowski, L., & Ptacek, L. J. (1999). Familial advanced sleep-phase syndrome:Ashort-period circadian rhythm variant in humans. Nature Medicine, 5(9), 1062–1065.
- Kerkhof, G. A., Van Dongen, H. P., & Bobbert, A. C. (1998). Absence of endogenous circadian rhythmicity in blood pressure? American Journal of Hypertension, 11, 373–377.
- King, D. P., & Takahashi, J. S. (2000). Molecular genetics of circadian rhythms in mammals. Annual Review of Neuroscience, 23, 713–742.
- Knutsson, A., Akerstedt, T., Jonsson, B. G., & Orth-Gomer, K. (1986). Increased risk of ischaemic heart disease in shift workers. Lancet, 2(8498), 89–92.
- Kolker, D. E., & Turek, F. W. (2001). Circadian rhythms and sleep in aging rodents. In P. R. Hof & C. V. Mobbs (Eds.), Functional neurobiology of aging (pp. 869–882). San Diego, CA: Academic Press.
- Landolt, H. P., Dijk, D. J., Achermann, P., & Borbely, A. A. (1996). Effects of age on the sleep EEG: Slow-wave activity and spindle frequency in young and middle-aged men. Brain Research, 738, 205–212.
- Leproult, R., Copinschi, G., Buxton, O., & Van Cauter, E. (1997). Sleep loss results in an elevation of cortisol levels the next evening. Sleep, 20(10), 865–870.
- Leproult, R., Van Reeth, O., Byrne, M. M., Sturis, J., & Van Cauter, E. (1997). Sleepiness, performance and neuroendocrine function during sleep deprivation: Effects of exposure to bright light or exercise. Journal of Biological Rhythms, 12, 245–258.
- Lowrey, P. L., Shimomura, K., Antoch, M. P., Yamazaki, S., Zemenides, P. D., Ralph, M. R., Menaker, M., & Takahashi, J. S. (2000). Positional syntenic cloning and functional characterization of the mammalian circadian mutation tau. Science, 288(5465), 483–492.
- McEwen, B. S., & Sapolsky, R. M. (1995). Stress and cognitive function. Current Opinions in Neurobiology, 5, 205–216.
- McGinty, D., & Stern, N. (1988). Circadian and sleep-related modulation of hormone levels: Changes with aging. In J. R. Sowers & J. V. Felicetta (Eds.), Endocrinology of aging (pp. 75–111). New York: Raven Press.
- Mendelson, W. B., & Bergmann, B. M. (1999). EEG delta power during sleep in young and old rats. Neurobiology of Aging, 20(6), 669–673.
- Monk, T. H., Buysse, D. J., Reynolds, C. F., Berga, S. L., Jarrett, D. B., Begley, A. E., & Kupfer, D. J. (1997). Circadian rhythms in human performance and mood under constant conditions. Journal of Sleep Research, 6, 9–18.
- Monk, T. H., Buysse, D. J., Reynolds, C. F., Jarrett, D. B., & Kupfer, D. J. (1992). Rhythmic vs. homeostatic influences on mood, activation, and performance in young and old men. Journal of Gerontology: Psychological Sciences, 47, 221–227.
- Monk, T. H., Moline, M. L., Fookson, J. E., & Peetz, S. M. (1989). Circadian determinants of subjective alertness. Journal of Biological Rhythms, 4, 393–404.
- Moore, R. Y. (1991). Development of the suprachiasmatic nucleus. In D. C. Klein, R. Y. Moore, & S. M. Reppert (Eds.), Suprachiasmatic nucleus: The mind’s clock (pp. 391–404). New York: Oxford University Press.
- Mrosovsky, N. (1996). Locomotor activity and non-photic influences on the circadian clock. Biological Reviews, 71, 343–372.
- Naylor, E., Penev, P., Orbeta, L., Janssen, I., Ortiz, R., Colecchia, E. F., Keng, M., Finkel, S., & Zee, P. C. (2000). Daily social and physical activity increases slow-wave sleep and daytime neurophychological performance in the elderly. Sleep, 23(1), 87–95.
- Penev, P., Zee, P. C., Wallen, E. P., & Turek, F. W. (1995). Aging alters the phase-resetting properties of a serotonin agonist on hamster circadian rhythmicity. American Journal of Physiology, 268, R293–R298.
- Porkka-Heiskanen, T., Strecker, R. E., Thakkar, M., Bjorkum, A. A., Greene, R. W., & McCarley, R. W. (1997). Adenosine: A mediator of the sleep-inducing effects of prolonged wakefulness. Science, 276, 1255–1258.
- Prinz, P. N. (1995). Sleep and sleep disorders in older adults. The Journal of Clinical Neurophysiology, 12, 139–146.
- Prinz, P. N., Peskind, E. R., Vitaliano, P. P., Raskind, M. A., Eisdorfer, C., Zemcuznikov, N., & Gerber, C. J. (1982). Changes in the sleep and waking EEGs of nondemented and demented elderly subjects. Journal of American Geriatric Society, 30, 86–93.
- Ralph, M., Foster, R. G., Davis, F. C., & Menaker, M. (1990). Transplanted suprachiasmatic nucleus determines circadian period. Science, 247, 975–978.
- Rechtschaffen, A., & Kales, A. (1968). A manual of standardized terminology, techniques and scoring system for sleep stages of human subjects. Los Angeles, CA: UCLA Brain Information Service/Brain Research Institute.
- Rechtschaffen, A., & Siegel, J. (2000). Sleep and dreaming. In E. R. Kandel, J. H. Schwartz, & T. M. Jessell (Eds.), Principles of neural science (4th ed., pp. 936–947). New York: McGraw-Hill.
- Reid, K. J., Chang, A. M., Dubocovich, M. L., Turek, F. W., Takahashi, J. S., & Zee, P. C. (2001). Familial advanced sleep phase syndrome. Archives of Neurology, 58(7), 1089–1094.
- Reppert, S. M., & Weaver, D. R. (1991). A biological clock is oscillating in the fetal suprachiasmatic nuclei. In D. C. Klein, R. Y. Moore, & S. M. Reppert (Eds.), Suprachiasmatic nucleus: The mind’s clock (pp. 405–418). New York: Oxford University
- Rosenberg, R. S., Zepelin, H., & Rechtschaffen, A. (1979). Sleep in young and old rats. Journal of Gerontology, 34(4), 525–532.
- Rosenthal, N. E., Joseph-Vanderpool, J. R., Levendosky, A. A., Johnston, S. H., Allen, R., Kelly, K. A., Souetre, E., Schultz, P. M., & Starz, K. E. (1990). Phase-shifting effects of bright morning light as treatment for delayed sleep phase syndrome. Sleep, 13(4), 354–361.
- Roth, T., & Roehrs, T. (2000). Sleep organization and regulation. Neurology, 54(5), S2–S7.
- Sack, R. L., Hughes, R. J., Edgar, D. M., & Lewy, A. J. (1997). Sleep-promoting effects of melatonin: At what dose, in whom, under what conditions and by what mechanisms? Sleep, 20, 908–915.
- Saini, J., Krieger, J., Brandenberger, G., Wittersheim, G., Simon, C., & Follenius, M. (1993). Continuous positive airway pressure treatment: Effects on growth hormone, insulin and glucose profiles in obstructive sleep apnea patients. Hormone and Metabolic Research, 25(7), 375–381.
- Sancar, A. (2000). Cryptochrome: the second photoactive pigment in the eye and its role in circadian photoreception. Annual Review of Biochemistry, 69, 31–67.
- Sanford, J. R. (1975). Tolerance of debility in elderly dependents by supporters at home: its significance for hospital practice. British Medical Journal, 3, 471–473.
- Satinoff, E., Li, H., Tcheng, T. K., Liu, C., McArthur, A. J., Medanic, M., & Gillette, M. U. (1993). Do the suprachiasmatic nuclei oscillate in old rats as they do in young ones? American Journal of Physiology, 265, R1216–R1222.
- Sherin, J. E., Shiromani, P. J., McCarley, R. W., & Saper, C. B. (1996). Activation of ventrolateral preoptic neurons during sleep. Science, 271, 216–219.
- Siegel, J. M. (1995). Phylogeny and the function of REM sleep. Behavioural Brain Research, 69(9), 29–34.
- Siegel, J. M. (1999). The evolution of REM sleep. In R. Lydic & B. Baghdoyan (Eds.), Handbook of behavioral state control (pp. 87–100). Boca Raton, FL: CRC Press.
- Spiegel, K., Leproult, R., Colecchia, E., L’Hermite-Balériaux, M., Zhiqun, N., Copinschi, G., & Van Cauter, E. (2000). Adaptation of the 24-hour growth hormone profile to a state of sleep debt. American Journal of Physiology (Endocrinology and Metabolism), 279(3), R874–R883.
- Spiegel, K., Leproult, R., & Van Cauter, E. (1999). Impact of sleep debt on metabolic and endocrine function. Lancet, 354, 1435–1439.
- Stepanski, E., Zorick, F., Roehrs, T., & Roth, T. (2000). Effects of sleep deprivation on daytime sleepiness in primary insomnia. Sleep, 23(2), 215–219.
- Thomas, M., Sing, H., Belenky, G., Holcomb, H., Mayberg, H., Dannals, R., Wagner, H., Thorne, D., Popp, K., Rowland, L., Welsh, A., Balwinski, S., & Redmond, D. (2000). Neural basis of alertness and cognitive performance impairments during sleepiness: I. Effects of 24 h of sleep deprivation on waking human regional brain activity. Journal of Sleep Research, 9(4), 335–352.
- Tobler, I. (2000). Phylogeny of sleep regulation. In M. H. Kryger, T. Roth, & W. C. Dement (Eds.), Principles and practices of sleep medicine (pp. 72–81). Philadelphia: W. B. Saunders.
- Toh, K. L., Jones, C. R., He, Y., Eide, E. J., Hinz, W. A., Virshup, D. M., Ptacek, L. J., & Fu, Y. H. (2001). An hPer2 phosphorylation site mutation in familial advanced sleep phase syndrome. Science, 291(5506), 1040–1043.
- Turek, F. W. (1998). Circadian rhythms. Hormone Research, 49, 103–113.
- Turek, F. W., & Van Reeth, O. (1996). Circadian Rhythms. In M. J. Fregly & C. M. Blatteis (Eds.), Handbook of physiology: Environmental physiology (pp. 1329–1360). Oxford, UK: Oxford University Press.
- Van Cauter, E., Blackman, J. D., Roland, D., Spire, J. P., Refetoff, S., & Polonsky, K. S. (1991). Modulation of glucose regulation and insulin secretion by circadian rhythmicity and sleep. The Journal of Clinical Investigation, 88, 934–942.
- Van Cauter, E., Copinschi, G., & Turek, F. W. (2001). Endocrine and other biologic rhythms. In K. L. Becker (Ed.), Endocrinology (4th ed., pp. 235–256). Philadelphia: W. B. Saunders.
- Van Cauter, E., Leproult, R., & Plat, L. (2000). Age-related changes in slow-wave sleep and REM sleep and relationship with growth hormone and cortisol levels in healthy men. The Journal of the American Medical Association, 284, 861–868.
- Van Cauter, E., Plat, L., & Copinschi, G. (1998). Interrelations between sleep and the somatotropic axis. Sleep, 21, 553–566.
- Van Cauter, E., Polonsky, K. S., & Scheen, A. J. (1997). Roles of circadian rhythmicity and sleep in human glucose regulation. Endocrine Reviews, 18(5), 716–738.
- Van Cauter, E., Sturis, J., Byrne, M. M., Blackman, J. D., Leproult, , Ofek, G., L’Hermite-Balériaux, M., Refetoff, S., Turek, F. W., & Van Reeth, O. (1994). Demonstration of rapid lightinduced advances and delays of the human circadian clock using hormonal phase markers. American Journal of Physiology, 266, E953–E963.
- Van Cauter, E., & Turek, F. W. (1995). Endocrine and other biological rhythms. In L. J. DeGroot (Ed.), Endocrinology (pp. 2487–2548). Philadelphia: W. B. Saunders.
- Van Cauter, E., & Turek, F. W. (2001). Roles of sleep-wake and dark-light cycles in the control of endocrine, metabolic, cardiovascular and cognitive function. In B. S. McEwen (Ed.), Handbook of physiology: Vol. 4. Coping with the environment: Neural and endocrine mechanisms (pp. 313–330). New York: Oxford University Press.
- van Coevorden, A., Mockel, J., Laurent, E., Kerkhofs, M., L’Hermite-Balériaux, M., Decoster, C., Nève, P., & Van Cauter, E. (1991). Neuroendocrine rhythms and sleep in aging men. American Journal of Physiology, 260, E651–E661.
- Van Dongen, H. P., Maislin, G., & Kerkhof, G. A. (2001). Repeated assessment of the endogenous 24-hour profile of blood pressure under constant routine. Chronobiology International, 18(1), 85–98.
- Van Gool, W. A., & Mirmiran, M. (1986). Aging and circadian rhythms. In D. F. Swaab, E. Fliers, M. Mirmiran,W.A.Van Gool, & F. Van Haaren (Eds.), Aging of the brain and Alzheimer’s disease (Vol. 70, pp. 255–278).Amsterdam: Elsevier.
- VanHelder, T., Symons, J. D., & Radomski, M. W. (1993). Effects of sleep deprivation and exercise on glucose tolerance. Aviation, Space and Environmental Medicine, 64(6), 487–492.
- Van Reeth, O., Sturis, J., Byrne, M. M., Blackman, J. D., L’HermiteBalériaux, M., Leproult, R., Oliner, C., Refetoff, S., Turek, F. W., & Van Cauter, E. (1994). Nocturnal exercise phase delays circadian rhythms of melatonin and thyrotropin in normal men. American Journal of Physiology (Endocrinology and Metabolism), 266, E964–E974.
- Van Reeth, O., Zhang, Y., Reddy, A., Zee, P. C., & Turek, F. W. (1993). Aging alters the entraining effects of an activity-inducing stimulus on the circadian clock. Brain Research, 607, 286–292.
- Van Reeth, O., Zhang, Y., Zee, P. C., & Turel, F. W. (1992). Aging alters feedback effects of the activity-rest cycle in the circadian clock. American Journal of Physiology, 263, R981–R986.
- Vgontzas, A. N., Bixler, E. O., Lin, H. M., Prolo, P., Mastorakos, G., Vela-Bueno, A., Kales, A., & Chrousos, G. P. (2001). Chronic insomnia is associated with nyctohemeral activation of the hypothalamic-pituitary-adrenal axis: Clinical implications. Journal of Clinical Endocrinology and Metabolism, 86(8), 3787–3794.
- Waldstreicher, J., Duffy, J. F., Brown, E. N., Rogacz, S., Allan, J. S., & Czeisler, C. A. (1996). Gender differences in the temporal organization of prolactin (PRL) secretion: Evidence for a sleep-independent circadian rhythm of circulating PRL levels. A Clinical Research Center study. Journal of Clinical Endocrinology and Metabolism, 81, 1483–1487.
- Wehr, T., Moul, D., Barbato, G., Giesen, H., Seidel, J., Barker, C., & Bender, C. (1993). Conservation of photoperiod-responsive mechanisms in humans. American Journal of Physiology, 265, R846–R857.
- Weibel, L., Follenius, M., Spiegel, K., Ehrhart, J., & Brandenberger, G. (1995). Comparative effect of night and daytime sleep on the 24-hour cortisol secretory profile. Sleep, 18, 549–556.
- Weibel, L., Spiegel, K., Follenius, M., Ehrhart, J., & Brandenberger, G. (1996). Internal dissociation of the circadian markers of the cortisol rhythm in night workers. American Journal of Physiology, 270(4, Pt. 1), E608–E613.
- Weitzman, E. D., Czeisler, C. A., Coleman, R. M., Spielman, A. J., Zimmerman, J. C., Dement, W., Richardson, G., & Pollak, C. P. (1981). Delayed sleep phase syndrome: A chronobiological disorder with sleep-onset insomnia. Archives of General Psychiatry, 38(7), 737–746.
- Weitzman, E. D., Zimmerman, J. C., Czeisler, C. A., & Ronda, J. M. (1983). Cortisol secretion is inhibited during sleep in normal man. Journal of Clinical Endocrinology and Metabolism, 56, 352–358.
- Welsh, D. K., Richardson, G. S., & Dement, W. C. (1986). Effect of age on the circadian pattern of sleep and wakefulness in the mouse. Journal of Gerontology, 41, 579–586.
- Wever, R. A. (1986). Use of bright light to treat jet lag: Differential effects of normal and bright artificial light on human circadian rhythms. Annals of the New York Academy of Sciences, 453, 282–304.
- Wise, P. M., Cohen, I. R., Weiland, N. G., & London, D. E. (1988). Aging alters the circadian rhythm of glucose utilization in the suprachiasmatic nucleus. Proceedings of the National Academy of Sciences, USA, 85, 5305–5309.
- Wright, K. P. J., Badia, P., Myers, B. L., Plenzler, S. C., & Hakel, M. (1997). Caffeine and light effects on nighttime melatonin and temperature levels in sleep-deprived humans. Brain Research, 747, 78–84.
- Zepelin, H. (2000). Mammalian sleep. In M. H. Kryger, T. Roth, & W. C. Dement (Eds.), Principles and practices of sleep medicine (pp. 82–92). Philadelphia: W. B. Saunders.
ORDER HIGH QUALITY CUSTOM PAPER

Always on-time

Plagiarism-Free

100% Confidentiality